WES助力伴胃弥漫性平滑肌瘤和肾病家族史16岁男性临床诊断,检出COL4A5/COL4A6基因变异确诊Alport综合征
时间:2026-05-05 17:37:34 热度:37.1℃ 作者:网络
Alport综合征(AS)是一种由COL4A3、COL4A4和COL4A5基因突变所致的遗传性肾病,罕见的COL4A5–COL4A6连续基因改变会引发伴弥漫性平滑肌瘤病的Alport综合征(AS-DL)。一名 16 岁男性自童年起便存在轻度蛋白尿、血尿、听力损失及近视症状,估算肾小球滤过率为 82.8 mL/min/1.73 m²,肾活检显示节段性系膜硬化,免疫荧光检测结果为阴性,电子显微镜检查可见肾小球基底膜弥漫性变薄、足细胞足突融合。该患者的两名已故兄弟均患有终末期肾病,其母亲存在血尿、子宫肌瘤及膀胱良性肿瘤病史,依据Flinter诊断标准,患者被确诊为X连锁Alport综合征,随即启动了肾保护治疗。2024 年起患者主诉出现上腹部不适症状,内镜检查发现一处 2 cm大小的胃黏膜下病变,全外显子组测序检出COL4A5基因半合子错义变异,同时存在COL4A6基因1-2外显子缺失,最终确诊为伴弥漫性平滑肌瘤病的Alport综合征。本病例呈现了伴弥漫性平滑肌瘤病的Alport综合征的共病特征,表明早期家系评估结合多学科框架下的临床-病理-遗传综合评估,可及时诊断非典型家族性伴弥漫性平滑肌瘤病的Alport综合征,改善临床管理效果。

背 景
Alport综合征(AS)是一类以进行性肾衰竭、感音神经性听力损失及眼部异常为特征的遗传性疾病。Alport综合征由编码IV型胶原的COL4A3、COL4A4或COL4A5基因致病变异引发,IV型胶原是维持肾小球基底膜完整性必不可少的结构组分。该病最常见的分型是由COL4A5基因变异导致的X连锁Alport综合征;COL4A3和COL4A4基因变异通常是常染色体隐性遗传型Alport综合征的致病基础。Alport综合征的患病率估计约为1/5000-1/10000,但由于确诊较晚、存在非典型表现,尤其是女性患者因莱昂化效应(也称X染色体失活)存在表型异质性,其实际疾病负担可能更高。部分X连锁Alport综合征患者会因携带包含COL4A5和COL4A6基因的连续缺失突变而并发弥漫性平滑肌瘤病(DL),相关报告显示该合并症的发生率约为 2%-5%。截至 2021 年,全球仅报道了约 30 个经基因检测确诊的Alport综合征伴弥漫性平滑肌瘤病(AS-DL)家系。弥漫性平滑肌瘤病在女性中更为多见,最常累及食管、气管及生殖道,需要多学科诊疗。
COL4A3或COL4A4基因杂合变异可能仅表现为单纯蛋白尿和局灶节段性肾小球硬化,无典型的眼耳症状,这往往会导致误诊。近年研究还显示,双基因遗传、嵌合变异及深度内含子变异也参与疾病发病,是导致疾病表型多样的原因。这些研究结果支持将扩展遗传检测纳入常规评估流程,尤其是临床表现不典型的情况下。近期一篇论文报道了一名X连锁Alport综合征患儿确诊食管弥漫性平滑肌瘤病的病例,凸显了将临床评估、组织学检查及家系研究相结合以早期发现肾外表现的价值。
本病例报告旨在描述一个罕见的Alport综合征合并弥漫性平滑肌瘤病的家族性病例,强调将家系评估与临床检查、肾脏组织学检查及基因检测相结合的诊断价值。
病 例
患者男,16 岁,因疑似Alport综合征,已在乌兹别克斯坦安集延地区多学科儿童医疗中心肾病科随访 11 年。患者自诉存在长期镜下血尿、轻度蛋白尿、进行性听力下降及近视症状。其母亲孕期在妊娠早期出现过妊娠中毒症;患者为足月分娩,出生体重 4200 g,母乳喂养至 3 岁。既往病史包括肺炎、复发性扁桃体炎及反复上呼吸道感染(每年发作 6-8 次)。家系图谱提示疾病为X连锁遗传(图1)。患者的两名兄长均在青少年时期因肾小球肾炎引发的终末期肾病去世。患者母亲自青少年期就出现血尿,36 岁时因症状性子宫肌瘤药物治疗无效接受了手术治疗,在先证者接受评估的 3 年前,母亲被查出患有膀胱良性肿瘤(组织学亚型信息缺失)。当时医生建议她接受化疗,化疗后其血尿症状加重。患者母亲未接受过Alport综合征相关基因检测,也无法获取外祖父母或母系兄弟姐妹的更多家系信息。
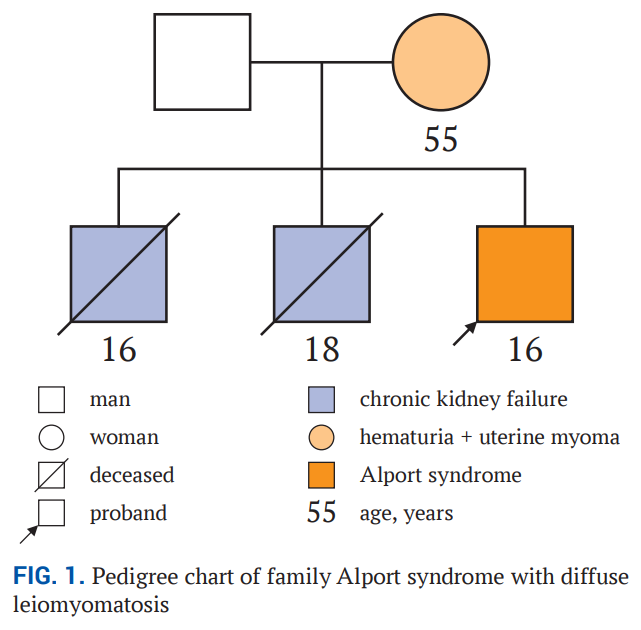
▲图1 伴有弥漫性平滑肌瘤病的Alport综合征家族的系谱图
2019 年,先证者接受了肾活检检查,共获取 3 条肾组织(总长度 37 mm),其中 1.5 mm的皮质片段留作电子显微镜检测使用。光学显微镜下可见系膜硬化样表现,伴节段性系膜硬化(图2A),间质纤维化累及约 10% 的肾皮质区域。免疫荧光检测未发现特异性肾小球沉积物(图2B),未开展针对淀粉样蛋白的刚果红染色。电子显微镜下可见肾小球基底膜弥漫性变薄,伴足细胞足突融合,符合Alport综合征的特征表现(图2C)。
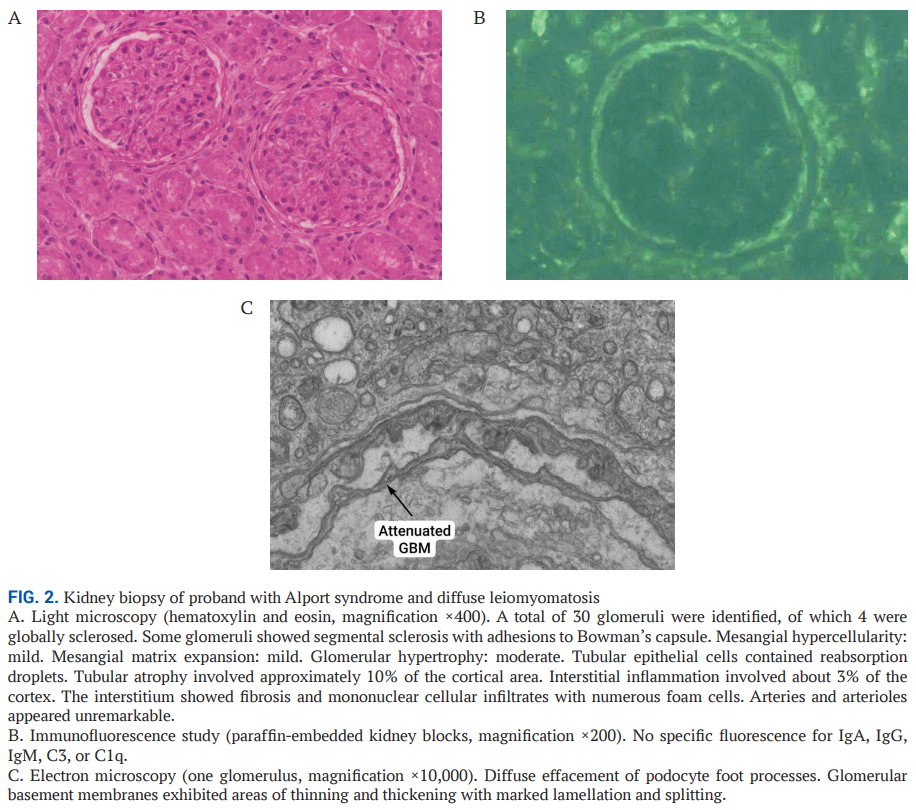
▲图2 Alport综合征和弥漫性平滑肌瘤病先证者的肾脏活检
心电图检查显示窦性心律,电轴正常。肾脏超声提示肾实质弥漫性改变,未见肾积水。眼科评估显示患者为II度近视,未记录到前圆锥形晶状体及点片状视网膜病变。听力测定显示双侧III度混合性听力损失(图3)。
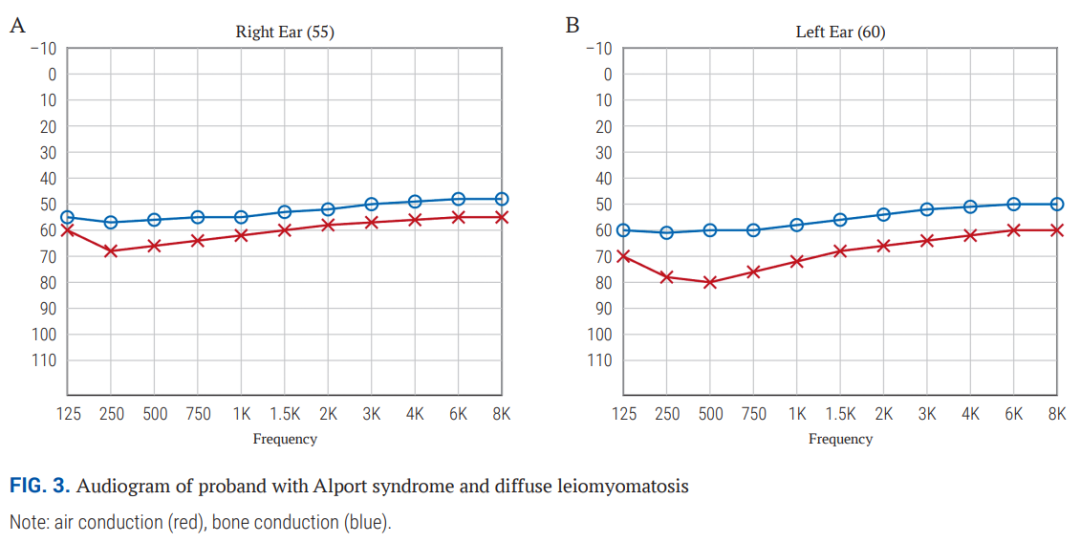
▲图3 Alport综合征和弥漫性平滑肌瘤病先证者的听力图
结合患者的临床表型(血尿、蛋白尿、混合性听力损失、近视)、组织病理学特征以及男性亲属存在青少年期肾衰竭的家族史,依据Flinter诊断标准,患者被确诊为X连锁Alport综合征。当时无法开展分子检测,随即启动了血管紧张素转换酶抑制剂类药物的肾保护治疗。2024 年起,患者主诉出现乏力、上腹痛、恶心、呕吐症状,心肺检查未发现异常。实验室检查提示中度贫血——血红蛋白 89 g/L,红细胞 3.6×10¹²/L,伴轻度白细胞减少(白细胞 3.9×10⁹/L)。血清生化检查显示总蛋白 54 g/L,肌酐 116 μmol/L,尿素 9.2 mmol/L,估算肾小球滤过率为 82.8 mL/min/1.73 m²。尿液分析显示尿蛋白 0.33 g/L,每高倍镜视野下白细胞 6-8 个,变形红细胞 10-12 个,均一形态红细胞 4-6 个,透明管型 2-5 个。2025 年 7 月,患者接受上消化道内镜检查,发现胃体部存在一处 2 cm大小的黏膜下病变(图4)。内镜下表现符合平滑肌肿瘤特征,建议行手术切除。在安排术前检查期间,患者经消化科与小儿外科联合随访,临床状态保持稳定。
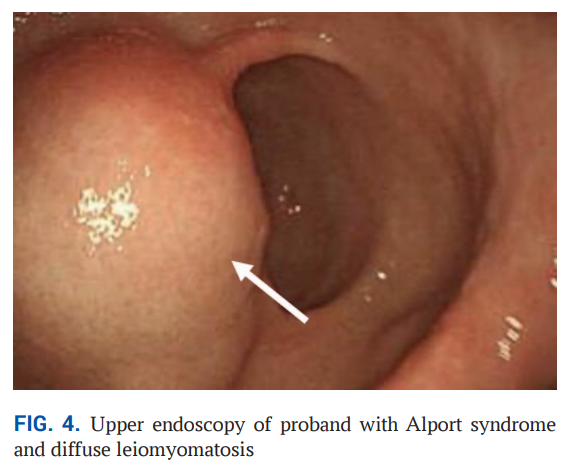
▲图4 患有Alport综合征和弥漫性平滑肌瘤病的先证者的上消化道内镜检查
鉴于患者的临床特征提示Alport综合征,同时检出符合弥漫性平滑肌瘤病表现的胃平滑肌肿瘤,有必要开展分子遗传学检测以明确诊断、指导临床规范管理。全外显子组测序检出两处致病变异:一处为COL4A5基因半合子错义变异(NM_000495.5:c.2879G>A, p.Gly960Asp),该变异会影响α5(IV)型胶原链,符合X连锁Alport综合征的致病特征;另一处为包含COL4A6基因1-2外显子的缺失(NM_000096.4),提示X染色体上存在COL4A5–COL4A6连续重排,这正是Alport综合征相关弥漫性平滑肌瘤病的致病基础。参考美国医学遗传学与基因组学学会指南,结合在线人类孟德尔遗传(OMIM)数据库的现有注释,这两处变异均被归类为致病变异。随着更多研究证据积累,该分类可能会有所调整。
讨 论
Alport综合征是一类由IV型胶原缺陷引发的进展性先天性肾病,典型病变累及肾脏、听觉系统及眼部,其中X连锁显性遗传型更为常见。本例患者早年就出现肾-耳受累表型,16 岁时出现上腹部症状,内镜检查发现胃体部有一处 2 cm大小的黏膜下病变,符合伴弥漫性平滑肌瘤病的Alport综合征中所见的平滑肌增生表现。家族史进一步支持X连锁遗传的推断:患者的两名兄长均在青少年时期因终末期肾病去世,母亲自青少年期起出现血尿,36 岁时因子宫肌瘤接受手术治疗,后续又检出膀胱良性肿瘤。这些临床特征提示患者可能罹患罕见类型的Alport综合征,潜在致病原因是COL4A5和COL4A6基因的微缺失。不过目前无论是临床层面还是遗传学层面,都尚未明确证实子宫肌瘤与Alport综合征存在直接关联。此外子宫肌瘤本身较为常见,在没有基因检测证实的情况下,不能直接假定其与Alport综合征存在因果关系。本病例也可能体现了Alport综合征的特点:相较于男性患者,女性患者的疾病表现更为轻微。
患者母亲的非特异性症状、两名兄长出现终末期肾病的结局,反映了遗传评估延迟导致的严重后果。家系特征支持X连锁遗传,同时也体现了该病多器官受累的异质性。肾脏、耳、眼、胃、子宫同时受累属于综合征性表现,凸显了多学科会诊的必要性。这些发现进一步印证了多学科管理在伴弥漫性平滑肌瘤病的Alport综合征诊疗中的价值,需要由肾病科医师、听力科医师、眼科医师及消化科医师定期随访,以实现疾病的早期发现、及时干预。
本例患者的临床病理特征吻合:肾活检显示节段性系膜硬化,伴 10% 的间质纤维化,以及肾小球基底膜典型的超微结构异常。免疫荧光检测未发现免疫球蛋白或补体沉积,提示该病属于基底膜结构异常疾病,而非免疫复合物介导的疾病。听力测定证实患者存在双侧III度混合性听力损失,眼科评估显示存在近视,符合Alport综合征已被认知的肾外表现谱。反复上呼吸道感染可能提示患者存在潜在的免疫功能异常,或与Alport综合征相关。
本例患者的肾功能相对保留(估算肾小球滤过率为 82.8 mL/min/1.73 m²),该结果提示本例Alport综合征的临床表型进展较慢,而非X连锁遗传型男性青少年患者中常见的肾功能快速下降。但Alport综合征通常呈进展性,仍有必要对患者进行严密监测。管理重点包括耐受情况下启用肾素-血管紧张素-醛固酮系统抑制剂、定期评估蛋白尿和肾小球滤过率,以及长期的听力学和眼科随访。
本病例也阐明了在分子检测资源有限的情况下可采取的诊断途径:结合家系分析、多器官评估及肾脏病理检查的结构化诊疗流程可以做出推定诊断,在条件允许时再安排患者接受基因检测明确诊断。对高危亲属开展遗传咨询可以作为分子检测的实用、高性价比替代方案,同时可依据Flinter标准明确诊断。
本病例展示了伴弥漫性平滑肌瘤病的X连锁Alport综合征,凸显了该病的多系统受累特征及家族性疾病负担。两名罹患慢性肾衰竭的兄长离世、母亲长期血尿合并平滑肌瘤病的情况,凸显了及时开展家系分析、采用多学科诊疗方案实现早期诊断的重要性。针对非典型遗传性疾病,家系评估与靶向基因检测能够帮助临床医师为患者及其亲属争取更优的疾病结局。
参考文献:
Boltaboeva, М & Haydarov, A. & Ganieva, M. & Kholmatov, D. & Efimenko, O. & Rakhmanova, L.. (2026). Familial co-occurrence of diffuse leiomyomatosis and Alport syndrome: a clinical case report. Sechenov Medical Journal. 16. 49-57. 10.47093/2218-7332.2025.16.4.1344.

