【AJH】克隆性造血(CH)与多种疾病的临床关联及CH的检测指征和干预策略
时间:2026-05-05 17:04:04 热度:37.1℃ 作者:网络
克隆性造血(CH)是指造血干细胞和祖细胞(hematopoietic stem and progenitor Cells,HSPC)中存在随时间推移导致克隆扩增的体细胞变异,潜质未定的克隆性造血(CHIP)在操作上定义为在变异等位基因频率≥2%的 HSPCs 中出现的致癌驱动基因致病性变异。
CH与进展性血细胞减少症(也称为不确定意义的克隆性血细胞减少症)、血液系统(主要是髓系,但也有淋巴系)肿瘤、细胞增多症(包括单核细胞增多症)以及非血液系统疾病(如动脉粥样硬化性心血管和脑血管疾病)风险增加相关。CH 还与许多其他疾病相关,包括静脉血栓栓塞、2 型糖尿病、慢性阻塞性肺疾病、骨质疏松症和痛风,并对阿尔茨海默病(AD)可能具有保护作用。
《American Journal of Hematology》近日发表综述,梅奥诊所Mrinal M. Patnaik教授等总结了 CH 的临床关联,并概述了 CH 的检测指征。

引言
克隆性造血(CH)定义为造血干细胞和祖细胞中存在导致克隆扩增的体细胞变异。CH 最初是通过观察健康女性髓系区室中随年龄增长而增加的非随机 X 染色体失活模式而发现的,随后被证实是继发于体细胞 TET2 突变,为未来涉及更大规模生物样本库和数据集的研究奠定了基础。有趣的是,CH 突变主要影响表观遗传调控基因(最常见的是:DNMT3A、TET2 和 ASXL1),这些基因也常见于髓系肿瘤,表明 CH 是一种肿瘤前驱事件,类似于浆细胞肿瘤中的单克隆丙种球蛋白病和慢性淋巴细胞白血病(CLL)中的单克隆 B 细胞淋巴细胞增多症;然而并非所有 CH 个体都会发展为髓系肿瘤。
最近的研究表明,CH 与多种恶性和非恶性疾病的关联正在不断演变。2014 年,对超过 17000 名个体的全外显子组测序研究确定,年龄 ≥40 岁的个体中 CH 频率很高;分别为 9.5%(70-79 岁)、11.7%(80-89 岁)和 18.4%(≥90 岁)。CH 的存在与全因死亡率(风险比 [HR] 1.4)、血液系统恶性肿瘤风险(HR 11.1)、缺血性冠状动脉疾病(HR 2.0)和中风(HR 2.6)增加相关;即使在调整了高龄、高体重指数、2 型糖尿病、性别和高血压等变量后,心血管事件仍与 CH 独立相关,随后的实验数据证实,在 TET2 敲除小鼠模型中,CH 与加速血管斑块形成存在因果关系。与之类似,将易患动脉粥样硬化的 LdIr−/−小鼠移植了 p53 缺陷的造血细胞后,发现其主动脉粥样硬化斑块大小和巨噬细胞积聚增加。
还有研究强调了 CH 与非恶性疾病(如 2 型糖尿病、骨质疏松症、慢性阻塞性肺疾病、痛风、静脉血栓慢性缺血性心力衰竭)的新关联。然而,由于缺乏前瞻性数据证明早期检测的临床效用以及干预策略有限,CH 检测的临床应用仍然有限。此外,由于 CH 在正常个体中普遍存在,对其过度医疗化是一个值得关注的问题,因为可能带来潜在的负面影响,例如增加焦虑、需要重复检测和/或骨髓活检等侵入性操作,以及因保险报销不确定性而增加的费用。
CH术语
由于最近关于 CH 的研究扩展,文献中术语的使用存在不一致。本文统一了与 CH 相关的术语,并提出可操作的定义(表 1 和图 1)。
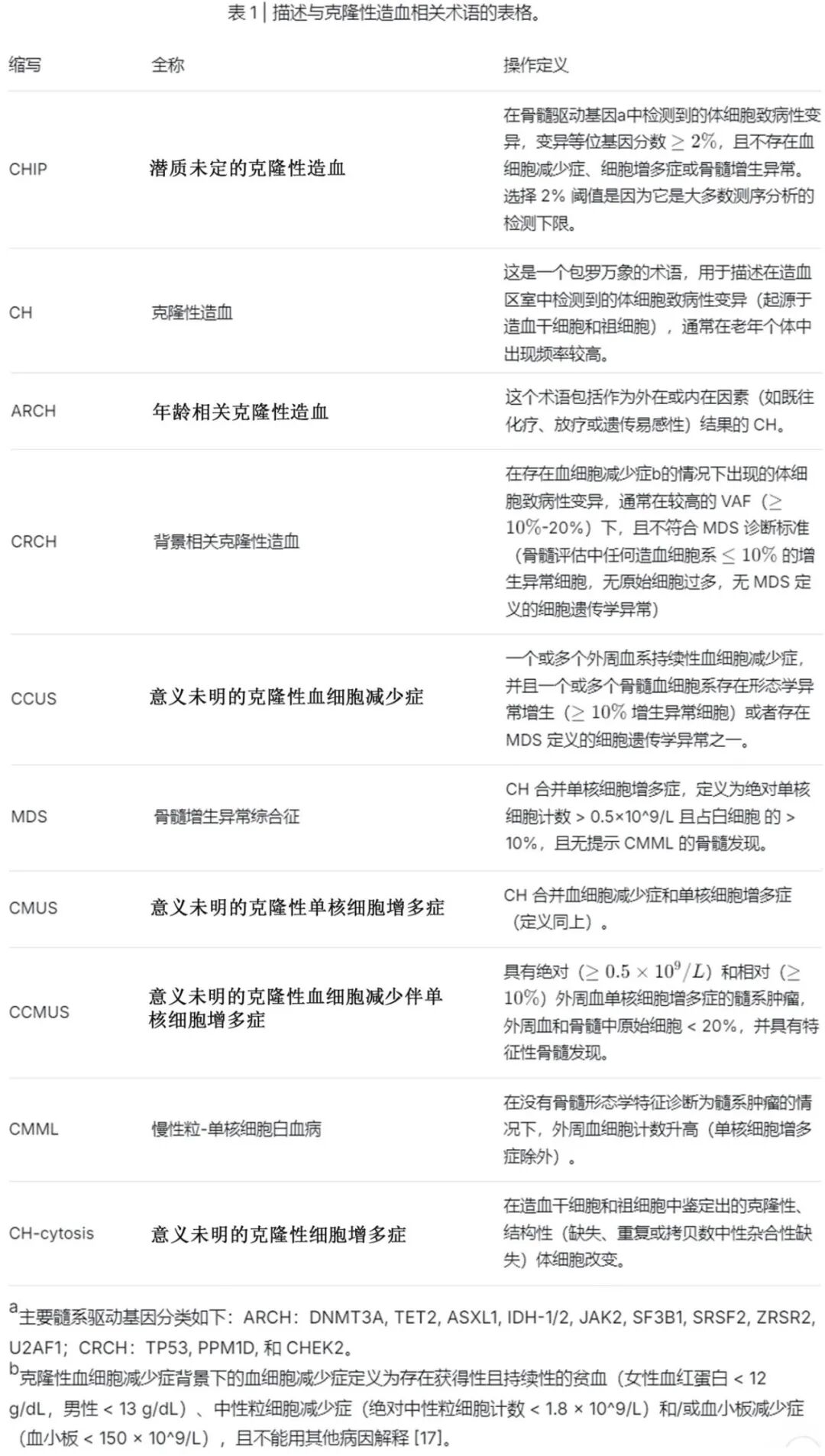
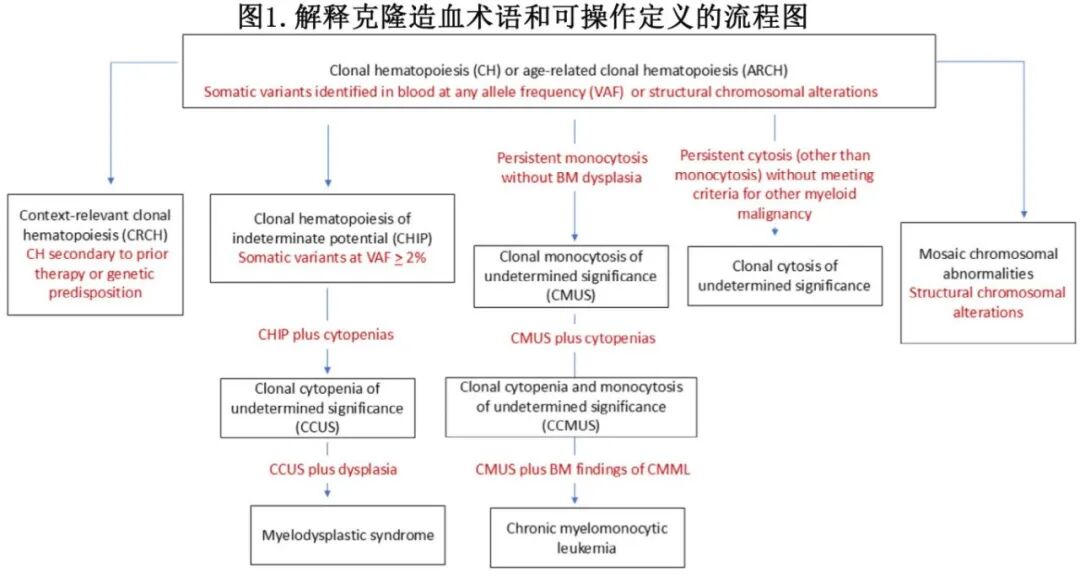
CH 也可称为年龄相关克隆性造血(ARCH),是一个包罗万象的术语,用于描述造血干细胞区室内致病性变异的发生。CH 可由点突变、插入/缺失或更大的结构重排驱动,后者特指嵌合染色体改变。且CH 可以根据病因、有无血细胞减少症或细胞增多症进一步细分。潜质未定的克隆性造血(CHIP)定义为在没有血细胞减少症、细胞增多症或骨髓增生异常的情况下,驱动癌基因中存在体细胞致病性变异(表 1),其变异等位基因频率(VAF)≥2%。VAF 低于 2% 的体细胞变异 CH 也可以通过更高灵敏度的纠错测序和深度靶向测序检测到。
到中年时,绝大多数个体都有低 VAF 的 CH 变异。纵向研究表明,这些变异大多保持静态甚至随时间缩小,表明只有一部分可能在以后的生命中具有临床意义(2% 阈值也是大多数靶向测序平台的检测下限)。根据病因,CH 可分为两大类:ARCH 和背景相关克隆性造血(context-relevant clonal hematopoiesis,CRCH)。若CH 个体出现持续≥4 个月的血细胞减少症(定义为存在获得性且持续性的贫血 [女性血红蛋白 < 12 g/dL,男性 < 13 g/dL]、中性粒细胞减少症 [绝对中性粒细胞计数 < 1.8 × 10^9/L] 和/或血小板减少症 [血小板 < 150 × 10^9/L],且不能用其他病因解释),则可以做出意义未明的克隆性血细胞减少症(CCUS)的诊断(图 1)。对于合并CH且持续性血细胞减少的患者,若其骨髓中有一个或多个谱系出现≥10%的细胞形态学发育异常,即可诊断为骨髓增生异常综合征或骨髓增生异常肿瘤(MDS)。
包含 MDS 定义分子事件的不断演变的分类,使得 MDS 和 CCUS 之间的区分更加复杂。在持续性绝对单核细胞增多的背景下,若单核细胞绝对计数 ≥ 0.5 × 10⁹/L,且占白细胞分类 ≥ 10%,但缺乏慢性粒-单核细胞白血病(CMML)的明确客观克隆及形态学标志,则称为“意义未明的克隆性单核细胞增多症”(clonal monocytosis of undetermined significance,CMUS);若同时合并血细胞减少,则称为“意义未明的克隆性血细胞减少伴单核细胞增多症”(clonal cytopenia and monocytosis of undetermined significance,CCMUS)。当外周血细胞计数升高(亦称“细胞增多症”)且缺乏诊断髓系肿瘤所需的骨髓形态学特征时,暂称为“意义未明的克隆性细胞增多症”(CH-cytosis)。
CH与血液系统恶性肿瘤
CH 与血液系统恶性肿瘤之间的关联突显了因果关系,因为 CH 变异是多种髓系和淋巴系统恶性肿瘤的早期启动事件。在 CH 变异等位基因分数>10% 的患者中(HR 49, 95% CI: 21–120),血液系统恶性肿瘤的风险增加了 11 倍(HR 11.1, 95% CI: 3.9–32.6)。CH 的患病率(70 岁以上个体 > 10%)与髓系恶性肿瘤的年发病率(每年 50,000–70,000 例)之间的差异表明,CH 突变获得并非白血病发生过程中的限速步骤。CH 突变特征、克隆复杂性、种系易感性以及细胞内在和外在因素在肿瘤发生中起着重要作用。
最近的研究表明,特定的 CH 驱动因素可能对髓系和淋巴系肿瘤显示出不同的风险。由髓系驱动基因突变驱动的"髓系"CH 强烈预示着髓系肿瘤风险,而由淋系驱动基因突变驱动的"淋系"CH 则与淋系肿瘤风险增加相关,最常见的是 CLL/SLL。然而这两个实体之间的重叠也越来越多,例如TET2 以前被认为是髓系特异性驱动基因,但也能促进 B 细胞淋巴系统肿瘤。此外,mCAs 的存在与淋巴系统恶性肿瘤而非髓系恶性肿瘤密切相关,可能反映出拷贝数事件作为淋巴系统恶性肿瘤早期驱动因素的重要性。髓系和淋系 CH 的已确立及候选基因列表见表 S1。

背景相关克隆性造血(CRCH)
治疗相关CH
研究强调了 CH与化疗和/或放疗(遗传毒性暴露)后发生治疗相关髓系肿瘤(t-MNs)之间的联系。在淋巴瘤患者中,任何 CH 的存在都与自体 HCT 后较差的总体生存期 和 t-MN 风险增加相关。正如预期,DNA 损伤反应和修复基因(如 PPM1D 和 TP53)的突变在这个队列中出现频率更高,可能是由于赋予了对烷化剂化疗和/或放疗的抵抗。PPM1D 基因编码一种丝氨酸/苏氨酸磷酸酶,负向调节 DDR 通路的多个组成部分(如 TP53、ATM、p16INK4a 和 ARP)。这些发现在一项丹麦研究中得到了证实,该研究也报告了涉及 DDR 通路基因(PPM1D、TP53、RAD21 和 BRCC3)的 CH 患者 OS 较差。然而,t-MN 风险增加不仅限于细胞毒性化疗。在多发性骨髓瘤患者中,CH 对 OS 的影响数据存在矛盾,一项研究报告了不利影响,而另一项则报告无影响,可能由具有免疫调节作用的来那度胺维持治疗(因此具有肿瘤保护作用)解释。
另一方面,由于选择性 CK1α(酪蛋白激酶)降解和随之而来的 p53 介导的 TP53 野生型细胞凋亡,TP53 突变克隆在暴露于来那度胺后显示出克隆优势。t-MN 在细胞毒性化疗治疗患者中发生率增加,是继发于先前存在的 CH 克隆扩增还是化疗诱导的遗传毒性应激,目前尚存争议,但越来越多的证据支持前一种假设。"背景"很重要(在这种情况下是暴露于烷化剂化疗和/或放疗),特别是在 TP53 突变克隆性血细胞减少症病例中,暴露于细胞毒性治疗(相对于未暴露)独立地导致更短的无进展生存期。然而,背景在"多打击"TP53 克隆性血细胞减少症(根据 ICC 分类,双等位基因受累,包括复杂核型、17p TP53 位点的 CN-LOH、两个不同突变且 VAFs≥10%,或单个突变且 VAF>50%)中是否具有同等重要性仍是一个悬而未决的问题。多打击 TP53 状态已确定为多种髓系疾病预后较差的独立预测因子。使用纠错测序等先进测序方法,已经在年轻个体中检测到 CH 克隆;甚至在脐带血造血干细胞中也有发现。
相当一部分(约 50% 的非霍奇金淋巴瘤和 MM 患者)在 CAR-T 细胞治疗前检测到 CH。与自体 HCT 不同,CAR-T 治疗前存在 CH 并不能预测不良 OS,但确实能预测更高的完全缓解率,以及在年轻(年龄 <60 岁)个体中发生 ≥2 级细胞因子释放综合征和神经毒性的风险。然而,有报道称,存在 CH(主要由先前的 TP53 和 PPM1D 突变克隆驱动)的患者CAR-T 细胞治疗后会发生 t-MN。当 TET2 基因通过慢病毒载体被基因破坏时,CAR T 细胞表现出向中央记忆表型转变的表观遗传特征,从而显示出增强的效力。然而,并非所有 CAR-T 后的恶性肿瘤都源于先前的 CH 克隆。CAR-T 输注后约 6% 的患者报告了继发性血液系统肿瘤(包括 T 细胞淋巴瘤),并且大部分是由于先前存在的 CH 突变和继发性事件,而非致癌性载体整合(有一例罕见的慢病毒整合 TP53 的病例例外)。CAR-T 后 t-MN 患病率增加在多大程度上反映了对先前细胞毒性治疗方案的暴露或 CAR-T 本身的影响,目前尚不清楚。
使用有 CH 的供者进行异基因 HCT 与慢性 GVHD 风险增加但复发率降低相关,对生存率没有总体影响。事实上,在移植时有疾病证据的受者中,使用 CH 供者(特别是 DNMT3A 突变)甚至可能是有利的,可能是由于更高的炎症相关的移植物抗白血病效应。供者 CH 的存在也与急性和慢性移植物抗宿主病发生率增加相关。移植后供者来源白血病的发生率很低,并且在 DTA(DNMT3A、TET2 或 ASXL1)突变的 CH 供体中未见,但当使用 TP53、剪接因子 CH 或种系 DDX41 突变供者时,出现过少数供者来源白血病的病例。目前没有足够的证据推荐在异基因 HCT 前对供者进行常规 CH 筛查。
CH与遗传性骨髓衰竭综合征
遗传性骨髓衰竭综合征(IBMFSs)包括一组具有多系统临床表现的遗传定义疾病,包括端粒长度维持基因(DKC1、TERT和TR)胚系突变引起的端粒生物学疾病 (TBDs)、DNA修复基因(FANCA-N基因)胚系突变引起的范可尼贫血(FA)、Shwachman—Diamond综合征 (SDS) 等。由于胚系突变造血干细胞固有的遗传缺陷,克隆演变和髓系肿瘤转化可发生在更早的年龄。CH 检测(特别是高危 CH,如存在剪接因子突变、TP53 和/或 VAF > 10% 的突变)具有临床价值,因其有助于决策是否进行异基因 HCT,尽管确切的进展率以及干预措施的性质和时机仍不清楚。这些疾病中 CH 发生较早的假设危,特定的遗传缺陷,如突变的 DNA 修复基因(FANCA-N)和突变的端粒维持基因(TERT、TR、DKC1、RTEL1),可导致受影响的 HSCs 存在固有的增殖劣势,这种劣势通过获得 CH 突变(适应不良的体细胞遗传补偿)得到补偿。在 SDS 中,既证明了通过 EIF6 双等位基因失活杂合突变增强克隆适应性(Fitness),也证明了通过 TP53 双等位基因突变进行适应不良补偿并随后转化为急性髓系白血病。
然而,获得增强的克隆适应性并不总是适应不良的(maladaptive),也可以保护其免于 MDS/AML 演变。在 TBDs 中,体细胞获得至少六种突变(TERT 启动子、POT1、TERF2IP、RBM7、SKIV2L2 和 DIS3)在无 MDS/AML 的患者中更为普遍,表明通过逆转通常与胚系 TBD 突变相关的端粒危机而起到保护机制的作用。相反,Chr1q+、U2AF1S34F和 TP53 突变在 TBD 中也更常见,其对未来肿瘤(MDS/AML)发展的风险不确定但可能增加。在 FA 中,伴有或不伴有染色体异常(+3q、-7q 和/或复杂核型)的 RUNX1 突变与适应不良补偿和肿瘤发展相关。可以考虑在 IBFMS 中进行 CH 检测,以回答治疗相关的问题,例如alloHSCT的时机,尽管由于缺乏前瞻性数据而尚未达成明确共识。
CH与遗传/胚系易感性
CH 的遗传易感性已在某些常见位点(如 TERT、SMC4、TCL1A、CD164、SETBP1、KPNA4-TRIM59、TET2、PARP1)以及罕见位点(如 ATM 和 CHEK2)的单核苷酸变异(SNV)和插入缺失中得到证实。英国生物样本库数据集发现,至少 8%-10% 的个体携带致病性胚系变异(PGV),其中绝大多数(> 99%)处于杂合状态。CHEK2(0.9%)最常见,其次是 ATM(0.5%)和 BRCA2(0.4%)。正如预期,这些携带者与年轻时发生 CH 有更强的关联,具有更大的最大 VAF 和突变数量,并且 mCA 风险也更高[最常见的是拷贝数中性杂合性缺失(cnLOH)],暗示了直接的因果关系。在 CH 突变中,DNMT3A 和 ASXL1 最常见,而在 CN LOH 中,1p、11q、13q 和 15q 是最常见的异常。一项全基因组关联研究报告了 TCL1A 启动子的遗传多态性,其减缓了 TET2、ASXL1、SF3B1 或 SRSF2 等驱动基因 CH 克隆的扩增速度,但这种保护作用在 DNMT3A 突变克隆中未观察到。DDX41 是一种相对常见的胚系易感基因,在大约 2%-5% 的髓系肿瘤病例中发生改变,可解释约 80% 的家族性 MDS 或 AML 病例。与预期相反,DDX41 胚系变异并不增加 CH 风险,从而突显了 MDS/AML 发展的不同机制。
表 S2 总结了所有已知 PGV 与 CH 的关联(表格过长,就不放图了)。
CH与免疫介导的再生障碍性贫血
据报道,大约 50% 的免疫介导的再生障碍性贫血患者存在 CH。不出所料,这种情况的一个显著特征危体细胞克隆(频率在 20% 到 30% 之间)占主导地位,这些克隆能逃避免疫攻击,例如 PIGA、BCOR、BCORL1 和 6p 的 CN-LOH/单亲二体性,并能预测对免疫抑制治疗和 OS 的良好反应。然而,传统 ARCH 基因(如 DNMT3A、TET2 和 ASXL1)的突变也以 5% 到 20% 的频率发生。然而,TP53、RUNX1 和剪接因子突变的存在预示着更高的 MDS/AML 发展风险,可以指导临床决策,例如异基因 HSCT 的时机。
CH与实体瘤
在一项大型回顾性研究中,大约 25% 的肿瘤患者检测到 CH,其中 4.5% 的 CH 突变发生在推定白血病驱动基因(putative leukemia driver genes,CH-PD)中。肿瘤中 CH 的危险因素包括年龄增长、既往接受放疗和吸烟;PPM1D 和 TP53 突变与既往接受化疗相关。在化疗暴露中,选择性药物如卡铂(ORs=1.4, p = 0.001)和拓扑异构酶 II 抑制剂(OR = 1.3, p = 0.01)与更高的 CH-PD 风险相关,而顺铂(OR = 1.1, p = 0.1)和奥沙利铂(OR = 0.98,p = 0.88)无影响。CH 改变经常在仅肿瘤标本中被识别出来,并且正如预期,在 60 岁以上的个体中更为常见。
a. CH 与实体瘤预后:PPM1D、CHEK2 和 TP53 突变预示着在接受肽受体放射性核素治疗(用于神经内分泌肿瘤的镥多肽)后转变为髓系肿瘤的可能性更高。类似地,TP53 突变克隆的存在与暴露于聚 ADP 核糖聚合酶抑制剂(PARP)(如芦卡帕利)后发生 t-MN 相关。最近,来自 TRACERx 队列的 421 名非小细胞肺癌(NSCLC)患者和来自 MSK-IMPACT 的 49351 名肿瘤患者的数据显示,肿瘤浸润性 CH 在所有癌症患者中观察到 26%(NSCLC 患者中 42%)。此外,肿瘤浸润性 CH 的存在与 TET2 突变密切相关,并被确定为较差的 OS 的独立预测因子 [HR 1.17 (95% CI 1.06–1.29)]。TET2 突变诱导的单核细胞迁移和随后对肿瘤微环境的重塑被认为是这些观察结果的机制。这些数据强调,CH 与微环境的变化相关,可能会产生正反馈循环并促进克隆进化。
然而,CH 并不总是预测实体瘤的不良结局。在转移性去势抵抗性前列腺癌中,CH 的存在不影响 OS,但较高的 VAF(≥10%)和 TET2 CH 与心血管事件增加相关。在转移性结肠癌中,DNMT3A CH 与改善 OS 相关(中位 OS 51.1 vs. 33.2 个月,p=0.044),可能是由于细胞毒性化疗后抗肿瘤免疫抑制活性增强。
b. CH 在循环肿瘤 DNA 检测中的作用:游离/循环肿瘤 DNA 检测可用于评估治疗资格(例如针对 DNA 修复基因突变的 PARP 抑制剂);然而大约 10%–40% 的样本存在 CH 变异。一项针对 465 名转移性结直肠癌和胰腺癌患者的大型研究显示,虽然 CH 很常见(10%–30%),但并不能预测化疗或免疫治疗后的不良风险。在解释此类检测结果时必须谨慎。突变类型(例如衰老相关特征 C>T)以及并发肿瘤加外周血突变筛查有助于区分真正的肿瘤变异与 CH 变异。
CH与心血管疾病
在分析了大型基于人群队列的样本后,确定了 CH 与心血管疾病之间存在显著且独立的关联。具体而言,在多变量模型中,CH 与缺血性心脏病(HR 2.0, p=0.015)和缺血性卒中(HR 2.6, p=0.003)相关。随后,两个独立的研究小组使用动物模型证实了这种关联具有因果关系;接受来自 Tet2−/− 敲除或 Jak2 V617F 条件性敲入小鼠的骨髓移植的易动脉粥样硬化 Ldlr−/− 小鼠具有更大的动脉粥样硬化病变,并且 TET2 缺陷的巨噬细胞表现出 NLRP3 炎症小体介导的白细胞介素-1β 分泌增加。个体而言,DNMT3A (HR 1.7, p=0.01)、TET2 (HR 1.9, p=0.06)、ASXL1 (HR 2.0, p=0.05)、JAK2 (HR 12, p<0.0001) 和"其他"组 (HR 2.2, p=0.002) 的突变与缺血性心脏病相关。CH 突变(TET2 或 DNMT3A, HR 2.1, p=0.02)也与缺血性心力衰竭相关,并存在不利的剂量关系,即较高的变异等位基因频率(≥10%)与不良结局相关。
然而,也有研究发现 CH 与心血管疾病之间没有关联或关联很弱。Kessler 等人在英国生物样本库的 628388 名个体中发现 CH 携带者的心血管疾病风险显著增加 (HR 1.11, 95% CI 1.03–1.19),但这主要是由 TET2 CH (HR 1.31, 95% CI 1.14–1.51) 驱动的。Kar 等人发现 CH 与缺血性心血管疾病(包括冠状动脉疾病和中风)之间没有显著关联。
这些矛盾的发现至少可以部分归因于抽样偏倚和 CH 检测标准的不同。最近,PESA 研究通过对 3692 名中年(40-55 岁)个体进行高灵敏度靶向测序和标准化非侵入性血管成像的纵向分析,显示了 CH 与新发股动脉粥样硬化发展之间的直接因果关系。证据的权重倾向于支持 CH 在促进动脉粥样硬化中的因果作用。
除了动脉粥样硬化,CH 还与慢性心力衰竭相关。从 DNMT3A CH 和慢性心力衰竭患者获得的外周血单核细胞转录组谱显示,炎症基因(如 IL1β、IL6、IL8、NLRP3 和巨噬细胞炎性蛋白 CCL3、CCL4,以及几个 T 细胞刺激基因)显著上调。类似地,系统性细胞因子谱分析研究显示,CH 患者中 IL-6、TNF-α 和 IL-18 等炎症细胞因子上调,这些因子可以抑制心肌功能。Bick 等人也报告了 IL6R rs2228145-C 多态性(这是 IL-6R 抑制的遗传代理指标)的心脏保护作用,这在一些研究中得到证实,但并非所有研究都如此,可能是由于 CH 过滤算法的差异。
CH 与炎症基因激活的关联不仅假设了加速动脉粥样硬化的因果机制,也提供了治疗干预的机会。CANTOS 试验将既往有心肌梗死且高敏 C 反应蛋白升高的患者随机分配接受卡那奴单抗(靶向 IL1β 的单克隆抗体)或安慰剂治疗,结果显示主要终点(非致死性心肌梗死、中风或心血管死亡)显著减少,一项探索性分析显示,在 TET2 突变 CH 患者中获益更大 [96]。此外,LoDoCo2 研究显示,与安慰剂相比,低剂量秋水仙碱(每天口服 0.5 mg)降低了心血管事件和死亡。同一研究的一项探索性亚组分析显示,低剂量秋水仙碱也可以减缓 TET2 突变 CH 的扩增。
CH与血液炎症性疾病
VEXAS 综合征是一种血液炎症综合征,伴有 UBA1 基因体细胞突变,引起一系列多系统临床特征,如多软骨炎、发热、骨髓衰竭、肺部炎症和单克隆丙种球蛋白病等。VEXAS 综合征患者中 DNMT3A 和 TET2 突变发生频率更高,数据表明它们共存预示着更高的死亡率,可能是由于更差的疾病表型。自 VEXAS 发现以来,一直在寻找其他可能激活自身炎症途径并产生炎症性全身环境的 CH 基因。有证据表明,IDH1 的体细胞突变可以引起类似表型。类似地,剪接基因如 SF3B1、SRSF2 和 U2AF1 也是激活炎症免疫信号传导并可能引起类似症状的候选基因。尽管有这些发现,仍然在许多具有"血液炎症"症状但没有遗传诊断的患者。这是一个新兴领域,预计未来会有许多这样的发现。
CH与非造血系统疾病
CH 与其他多种医学疾病相关。CH 突变和大染色体缺失的存在与 2 型糖尿病相关。类似地,CH(主要是 DNMT3A 和 TET2)也与慢性阻塞性肺疾病、肺癌、慢性肾脏疾病和痛风相关。CH 与中风风险增加相关;TET2 CH 与总体中风和缺血性中风相关,而 DNMT3A 与出血性中风风险增加相关。在缺血性卒中患者中,CH 还与白质病变负荷增加和促炎症谱相关,特别是 TET2 和 PPM1D 突变 CH 患者与继发性血管事件和缺血性卒中后死亡风险更高相关。DNMT3A CH 与骨质疏松症相关]。CH 与 COVID-19 发病率和死亡率的关系不一致,但与艰难梭菌和链球菌/肠球菌感染严重程度增加存在关联。有趣的是,DNMT3A 或 TET2 CH与阿尔茨海默病呈负相关。
表 2 总结了 CH 与非血液系统疾病的临床关联。
克隆适应性和选择压力
新的证据正在塑造对整个生命周期中驱动克隆选择和扩增因素的理解。CH 突变通常在晚年(年龄 >50 岁)被检测到,但通过灵敏检测(纠错测序),在年轻个体中也检测到较小的克隆。然而,VAF 小于 2% 的克隆随时间扩增的倾向较小;不过这在很大程度上取决于变异特异性的适应性影响和环境。特定的驱动突变(特别是 JAK2 V617F)据预测在子宫内就已发生,这是基于骨髓增殖性肿瘤中克隆的系统发育重建。如上所述,胚系易感性通常是大约 10% 个体较早发生 CH 的诱因。
大多数个体晚年 CH 扩增是由多种细胞内在和外在因素驱动的。CH 突变如 DNMT3A、TET2、ASXL1 和 JAK2 各自可提供不同的增殖优势,但也有一些 CH 突变(如剪接因子突变 SF3B1、SRSF2 和 U2AF1)具有竞争劣势(在体内模型中已证实),然而它们与克隆扩增相关。这表明存在其他变量,如吸烟、免疫选择、衰老相关炎症以及放化疗暴露,这些因素随着时间的推移优先促进克隆扩增。几项研究评估了哪些因素与 CH 扩增相关。
一项研究报告称,正选择(而非随机漂变)是 CH 的主要驱动力,并与 CH 突变赋予的不同程度的细胞适应性相关。几项研究指出,剪接突变如 SRSF2 P95R 和 SF3B1 K700E 具有最高的适应性,而非 R882H 的 DNMT3A 变异适应性较低,年增长率也较低。研究还评估了 AML 进展风险,发现 TP53 (HR 12.5, 95% CI 5–160.5)、U2AF1 (HR 7.9, 95% CI 4.1–192.2)、IDH(IDH1 和 IDH2, HR 28.5, 95% CI 2.5–879.1)和剪接基因(SF3B1, SRSF2 和 U2AF1, HR 7.4, 95% CI 1.7–32.2) CH 的效应量最高。在外部因素背景下,针对克隆特异性扩增的个体化模型可以为决策(例如进行辅助化疗和造血干细胞移植等)提供有价值的临床信息。
CH与临床预测模型
Weeks 等人开发了克隆性造血风险评分(CHRS),该评分使用临床/血液学(红细胞分布宽度、平均红细胞体积、血细胞减少症和年龄)和遗传变量(单一DNMT3A 突变、高危突变、突变数量和 VAF)将 CH 患者分为低危、中危和高危组,以预测其进展为髓系肿瘤的风险。每个变量被加权并分配分数:(A) 单一 DNMT3A 突变 0.5 分;(B) 无 DNMT3A 突变、无高危突变(SRSF2、SF3B1、ZRSR2、IDH1、IDH2、FLT3、RUNX1 和 JAK2 中的突变)、单一突变、VAF < 0.2、RDW < 15%、MCV < 100 fL、无血细胞减少症、年龄 < 65 岁各 1 分;(C) 存在血细胞减少症和年龄 ≥ 65 岁各 1.5 分;(D) 突变数量 ≥ 2、VAF ≥ 0.2 各 2 分;(E) 存在高危突变以及 RDW ≥ 15%、MCV ≥ 100 fL 各 2.5 分。将这些分数汇总为低危(<9.5)、中危(10-12)和高危(≥ 12.5)组,预测的 10 年髓系肿瘤累积发病率分别为 0.7%(±0.08)、7.8%(±0.8)和 52.2%(±5)。
Gu 等人报告了另一个名为"MN-predict"的模型(https://bioinf.stemcells.cam.ac.uk/shiny/vassiliou/MN_predict/),该模型结合临床(性别、血红蛋白、MCV、血小板计数)和遗传变量(CH 突变类型和 VAF),提供进展为 AML、MDS 和骨髓增殖性肿瘤的不同速率。Xie 等人提供了一个专门针对克隆性血细胞减少症患者的评分,称为"克隆性血细胞减少症风险评分",该评分确定了三个关键预后变量:存在剪接突变(2 分)、血小板计数 < 100 × 10^9/L(2.5 分)和 ≥ 2 个突变(3 分)。根据累积评分,患者被分为低危(<2.5 分)、中危(2.5 – < 5 分)和高危(≥ 5 分)组,预测的 2 年 MN 累积发病率分别为 6.4%、14.1% 和 37.2。
这些模型有助于为患者提供客观的进展风险;然而它们也有一定的局限性,例如未能纳入已被证明会影响 MN 风险的外在和内在变量,如既往接受化疗和/或放疗、吸烟暴露和胚系易感性。随着人工智能和机器学习等技术的出现,未来的模型有望实现个体化,并具有更优的预测能力。
检测建议
a. 持续≥4 个月不明原因的血细胞减少症:先前的研究表明,当对有不明原因血细胞减少症指征的个体进行 CH 检测时,大约 45%–60% 的患者被确定存在 CH 突变,也称为 CCUS。此外,VAF ≥10% 的体细胞突变、两个或更多突变、剪接因子基因之一存在突变以及与 DNMT3A、TET2 或 ASXL1 共存突变,对发展为髓系肿瘤具有非常高的阳性预测值 (0.8–1.0)。特定的突变,如 TP53 和 SF3B1,在贫血个体中富集。造血参数的其他异常,如红细胞分布宽度增加和白细胞增多,也可以预测 CH。尽管没有前瞻性证据表明诊断高危克隆性血细胞减少症后进行干预能改善结局,但有证据表明它可以改变预后判断和随访建议,因此对有此指征的个体进行筛查(图2)。
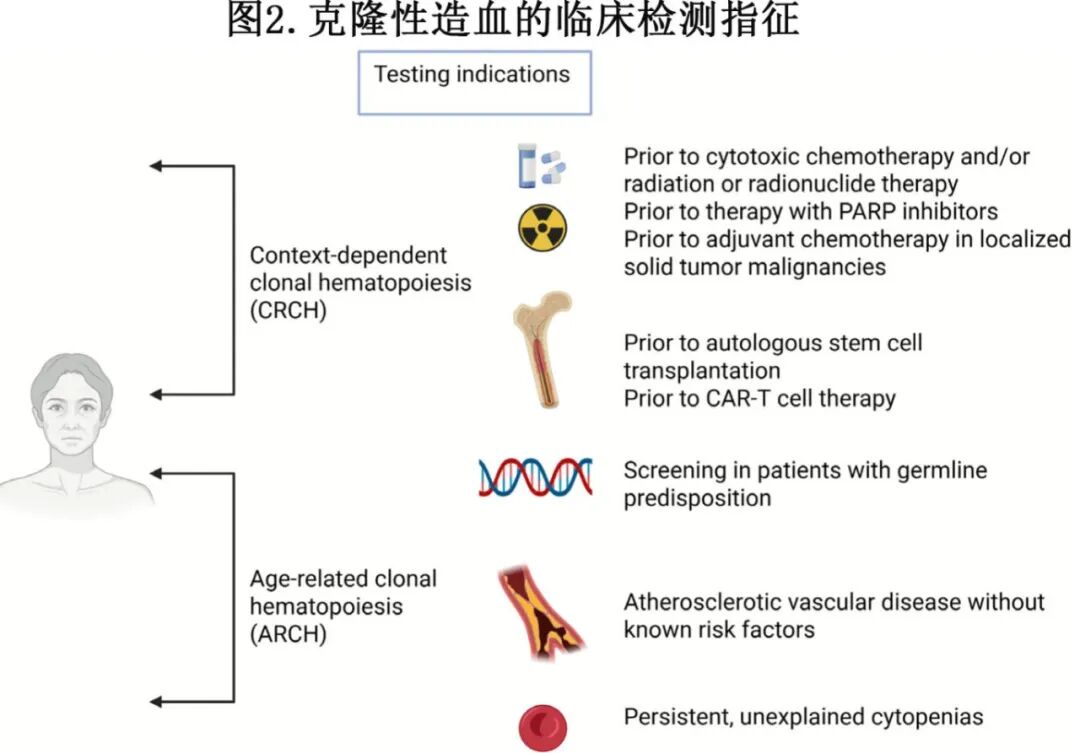
b. 在实体瘤测序或其他背景下的基因检测中偶然发现的 CH 突变:CH 相关改变可以在肺癌和乳腺癌等实体瘤测序过程中被发现。由于 CH 与内脏恶性肿瘤(例如 TP53、KRAS 和 EZH2)中观察到的体细胞突变存在重叠,应在外周血中确认它们的存在。游离/循环肿瘤 DNA 检测已被用于评估治疗资格(例如针对 DNA 修复基因突变的 PARP 抑制剂);然而,约 10%–40% 的样本存在 CH 变异。CH 的存在可以为这些患者未来的治疗策略提供信息;特别是,在评估辅助化疗或放疗前的风险与获益,以及在疾病进展时选择未来治疗方案时,应考虑这些信息。
c. 自体造血干细胞移植/CAR-T 前的筛查:如上所述,淋巴瘤和骨髓瘤患者在自体移植时存在 CH 预示着更差的总体生存期和无进展生存期。移植前的筛查还可以为患者未来的治疗选择提供信息,例如对于多发性骨髓瘤患者,来那度胺(而非泊马度胺)已被证明会促进 TP53 突变造血干细胞克隆的扩增。因此,建议对自体造血干细胞移植前的患者进行基于研究的 CH 筛查,特别是对于年龄 > 50 岁的个体,以及既往接受过化疗和/或放疗的患者。CAR-T 治疗前可以考虑 CH 筛查,但与自体 HCT 不同,目前尚无明确证据表明其对结局有影响。CAR-T 治疗前存在 CH 与不良 OS 无关(在有限随访范围内),但可能预测更高的反应率、≥ 2 级 CRS 风险更高,并且 TP53 CH 可能预测更高的髓系肿瘤风险 [38]。随着对关键 CAR-T 研究进行更长期随访数据的获得,先前存在的 CH 与结局(包括 t-MN)影响之间的关系将变得更加清晰。
d. PRRT、辅助化疗和 PARP 抑制剂治疗前的筛查:在治疗相对获益不明确,需要与发生 t-MN 的风险相权衡的情况下,应考虑在 PRRT、卵巢癌等适应症的辅助化疗和 PARP 抑制剂治疗前进行 CH 筛查。目前由于缺乏前瞻性数据,这些治疗后发展为 t-MN 的突变(和 VAF)特异性风险尚不清楚;然而,TP53 和可能 PPM1D 突变与 t-MN 之间的关联正在被确定,必须适当考虑。检测到 CH(即使是 TP53 和 PPM1D)并非这些治疗的绝对禁忌症,但必须在患者具体情况和替代(靶向和/或免疫)药物可用性的背景下仔细考虑。
e. 对患有动脉粥样硬化性血管疾病和缺血性心肌病且无法用常规危险因素解释的患者的筛查:累积证据不仅指向 CH 与动脉粥样硬化性血管疾病的相关性,而且指向其独立的因果关系。风险程度(HR 2.0)与传统危险因素如 2 型糖尿病(HR 3.1)和高血压(HR 2.3)相当。因此,对于动脉粥样硬化性血管疾病患者,特别是当缺乏传统危险因素时,可以考虑进行 CH 筛查(优先进行系统性细胞因子谱分析)。
f. 对有遗传易感性和 IBMFSs 的患者的筛查:建议对 IBMFS 患者进行选择性筛查,特别是在考虑复杂的治疗(如异基因 HCT)时。胚系嵌合变异有时会混淆结果,需要仔细检查。
干预策略
目前,对于 CH 患者尚无既定的干预策略。在作者的实践中,建议进行主动监测,每 3-6 个月监测一次全血细胞计数。对于具有两个以上 CH 突变,或剪接因子突变与 DTA 突变共存的患者,由于发展为髓系肿瘤的阳性预测值很高,建议进行密切的主动监测。在作者的 CH 门诊,还建议进行咨询,以控制可改变的风险因素,例如戒烟、通过生活方式改变和/或药物控制血脂异常和高血压。
目前正在进行一些针对克隆性血细胞减少症患者的临床试验,例如针对 IDH1 突变患者(NCT#05030441)的艾伏尼布(IDH1 抑制剂)多中心临床试验和针对 IDH2 突变患者(NCT#05102370)的 enasidenib(IDH2 抑制剂)治疗意义未明的克隆性血细胞减少症的临床试验。最近,在一项去中心化环境中进行的艾伏尼布研究显示,82% 的 IDH1 CCUS 患者出现血液学反应。静脉注射抗坏血酸可以恢复 TET2 缺陷的 TET2 突变造血干细胞,并逆转相关的干细胞更新缺陷。梅奥诊所进行了一项使用高剂量静脉注射抗坏血酸(1g/kg 体重,每周三次,持续 12 周)治疗 TET2 突变的克隆性血细胞减少症患者的初步研究(n=10,NCT#03418038),虽然没有观察到血液学反应或 TET2 VAF 的变化,但体外研究表明在关键的增强子区域发生了重要的表观遗传变化。
Canakinumab(靶向 IL-1β 的人单克隆抗体)也正在作为一种研究药物进行研究,以防止 CCUS 进展为明显的髓系肿瘤(NCT#05641831)。最近,对接受化疗加 CDK4/6 抑制剂试验患者的系列样本进行了分析。结果显示,CDK4/6 抑制剂暴露与 TP53 克隆生长减少相关,表明将传统化疗与 CDK4/6 抑制剂之一联合使用可能降低发生 TP53 突变 t-MN 或其前体状态的风险。其他靶点包括 NLRP3、IL-6、IL-18 和 TNF-α 等的抑制剂。
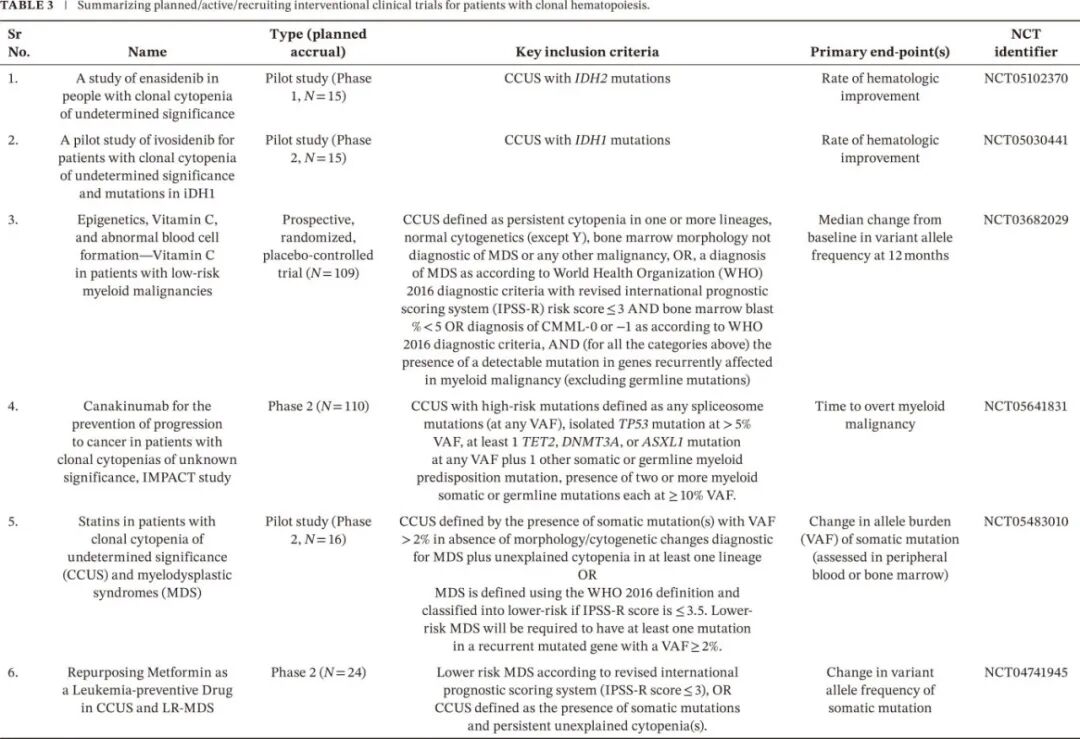
表 3 总结了 CH 的干预性研究,以及它们的纳入标准和主要终点。
未来方向
需要进行大型前瞻性研究来描绘 CH 进展的自然史,识别在外部因素背景下(如化疗和/或放疗暴露、炎症等)自然进展和扩增的真实突变特异性发生率。这将有助于精简监测指南,目标是早期识别肿瘤转化。有必要进行使用新型药物的早期干预试验,以评估是否能改变 CH 的自然史,特别是在那些即将发生进展的亚组中。CH 干预对非血液系统疾病的影响也需要进一步研究。
参考文献
Mangaonkar AA, Bolton KL, Patnaik MM. Clonal Hematopoiesis of Indeterminate Potential and Clonal Cytopenias of Undetermined Significance: 2026 Update on Clinical Associations and Management Recommendations. Am J Hematol. 2026 Jan 8. doi: 10.1002/ajh.70205. Epub ahead of print. PMID: 41508691.

