WES精准识别以急性脑病为特征的儿童「偏瘫型偏头痛」病因,助力早诊早治/改善预后
时间:2026-05-05 17:03:57 热度:37.1℃ 作者:网络
《头痛问诊要领中国专家建议(2025版)》指出:既往有肿瘤、免疫力低下(如获得性免疫缺陷综合征)、出血倾向、心血管病、精神障碍等疾病,均可为头痛的继发性因素。对于表现为偏头痛的患者,若有卒中、痴呆等家族史,应排除遗传性疾病,如1/3的伴皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL)患者早期可表现为有先兆的偏头痛样发作,头痛随着脑梗死、情绪及认知障碍的出现而逐渐缓解或消失;线粒体脑肌病伴乳酸酸中毒和卒中样发作(mitochondrial encephalopathy with lactic acidosis and stroke-like episodes,MELAS)患者可表现有或无先兆的偏头痛样发作,头痛出现于神经功能障碍之前或伴随发生。还需要询问物质、药物使用情况,以鉴别药物过度使用性头痛、酒精诱发的头痛、咖啡因戒断性头痛等,以及药物直接导致的头痛不良反应。

▲摘自《头痛问诊要领中国专家建议(2025版)》
偏瘫型偏头痛(HM)是一种罕见的偏头痛亚型。儿童偏瘫型偏头痛在疾病初期可能表现不典型,容易导致误诊。本文报道了 2 例以急性脑病起病的不典型偏瘫型偏头痛病例。通过PubMed、Web of Science和Scopus数据库进行了全面检索,仅纳入报道了携带CACNA1A或ATP1A2基因突变患者完整临床信息的文献。病例1的CACNA1A基因第5外显子存在新发突变c.674C>A(p.Pro225His),病例2的ATP1A2基因第16外显子存在错义突变(c.2143G>A,p.Gly715Arg)。结合我们的2例病例,最终共收集并总结了 160 例患者(73 例携带CACNA1A突变,87例携带ATP1A2突变)。急性脑病是儿童重度偏瘫型偏头痛发作的主要表现,这增加了诊断难度。对于出现嗜睡、昏迷或惊厥且无结构性、癫痫性、感染性或炎症性病因的患者,医师在鉴别诊断时应考虑偏瘫型偏头痛。当遇到类似临床病例时,基因检测尤为重要,有助于早期诊断和治疗。疾病的早期识别与治疗有助于改善预后。
背 景
偏瘫型偏头痛(HM)是一种罕见的偏头痛亚型,其特征为严重偏头痛发作及先兆症状,后者可累及单侧肢体,表现为运动无力或麻木(通常为轻偏瘫),还可伴随视觉、感觉或言语障碍。重度发作可伴随癫痫发作、昏迷、脑病、发热、小脑受累、脑水肿或脑梗死,且常被误诊为发作后意识模糊、托德瘫痪,或被诊断为癫痫、病毒性脑炎。本文报道了 2 例以急性脑病起病的不典型偏瘫型偏头痛病例。这 2 例患者的不典型临床表现给诊断带来了挑战。因此,本文报道该病例以加深对本病的认识,旨在减少误诊,为患者提供恰当有效的治疗,改善其生存质量。

▲摘自《中国偏头痛诊断与治疗指南(中华医学会神经病学分会第一版)》
病例1
患童男,3 岁,系非近亲父母的次子,足月顺产,母亲有偏头痛病史。因头痛、嗜睡、癫痫持续状态伴发热半天,入住青岛大学附属医院。患儿自1岁7个月起共出现 3 次类似发作,发病前后智力及运动发育均正常。每次发作前均有轻微头部外伤史,随后出现头痛、嗜睡及癫痫发作。癫痫发作表现为双眼向左凝视、左侧肢体间歇性强直伴意识丧失,且每次发作均为癫痫持续状态。本次癫痫发作缓解半天后,患儿出现发热,体温达 39℃。既往发作时头颅磁共振成像(MRI)、磁共振血管成像(MRA)及磁共振静脉成像(MRV)均未见异常;血常规、动脉血气分析、血氨、血糖、同型半胱氨酸、血钙及电解质均无明显异常。本次发作后头颅MRI显示,右侧额颞叶皮层在弥散加权成像(DWI)上呈多发高信号(图1)。本次发作后神经系统查体示:左侧中枢性面瘫,左侧上下肢瘫痪,双侧巴宾斯基征阳性,其余病理征阴性,脑膜刺激征阴性。
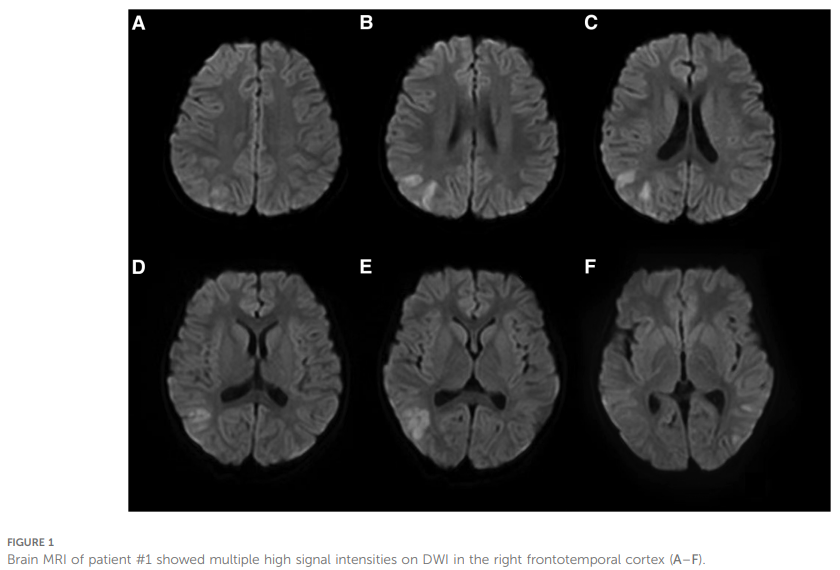
▲图1 病例1脑部MRI显示右侧额颞叶皮质DWI上存在多个高信号强度
脑脊液(CSF)检查显示,压力、葡萄糖、氯化物、蛋白水平、细胞计数及分类均正常;脑脊液细菌、真菌检测阴性。外周血及脑脊液脱髓鞘相关抗体(MOG、AQP4、MBP)均为阴性。视频脑电图(EEG)监测提示双侧半球持续不对称,右侧半球可见慢波及尖慢波发放,右侧半球生理波缺失(见图2);同期监测到1次局灶性发作,持续约 90 秒(见图3)。
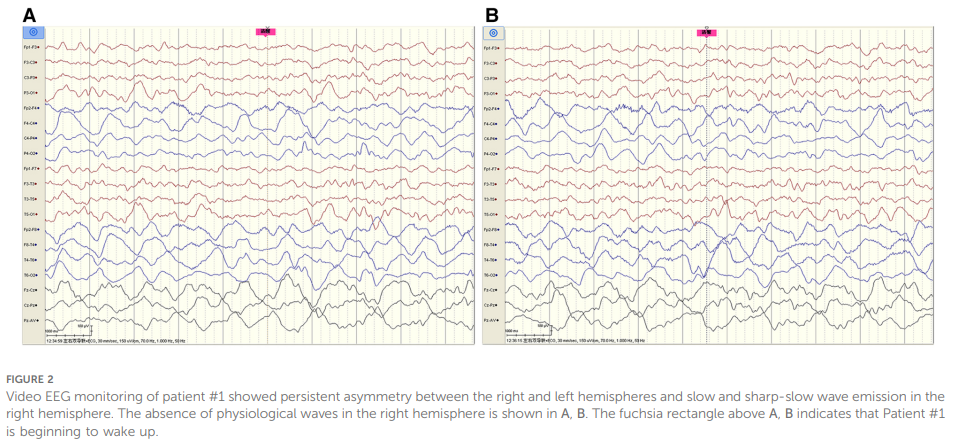
▲图2 病例1的脑电图监测显示,其左右半球持续存在不对称性,且右半球出现慢波和尖慢波
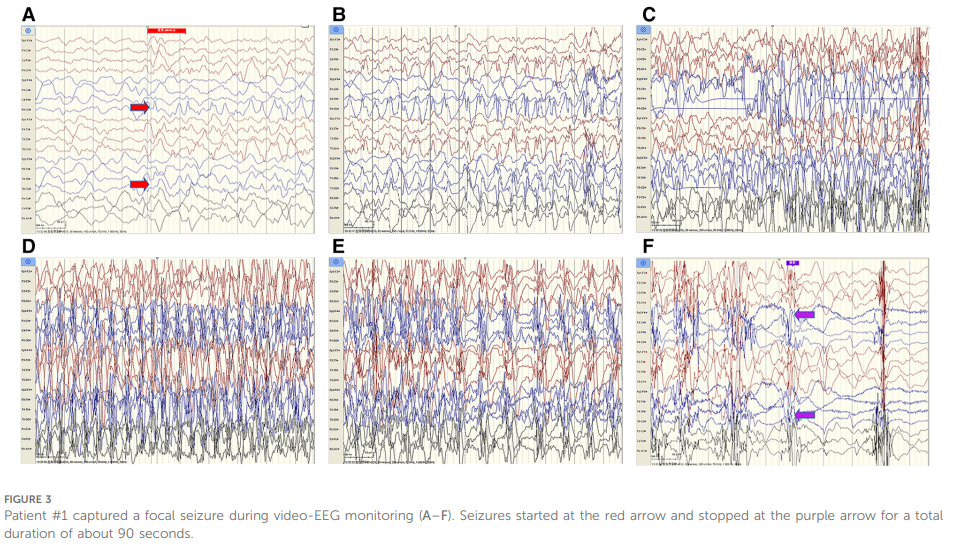
▲图3 病例1脑电图监测期间记录到一次局灶性癫痫发作
结合患者三次发作的临床表现,临床认为该患者极有可能是表现为急性脑病的偏瘫型偏头痛(HM)。除常规补液及营养支持治疗外,给予患者甲泼尼龙(3 毫克/千克/天)以减轻脑水肿,甘露醇(2.6 克/千克/天)以降低颅内压,同时给予氟桂利嗪(0.2 毫克/千克/天)扩张脑血管。患者意识障碍逐渐改善,左侧偏瘫也逐步缓解,发病 9 天后完全康复,未遗留后遗症。发病第 11 天复查视频脑电图显示,双侧枕区背景活动不对称,右侧为慢波覆盖,右侧半球以慢波、尖慢波发放为主(见图4)。
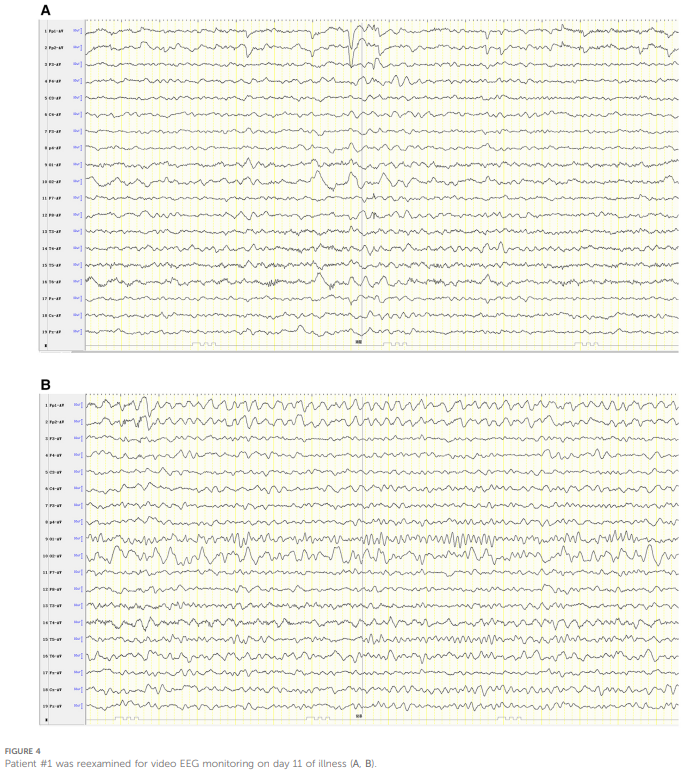
▲图4 病例1在发病第 11 天接受了脑电图监测复查
与患者家属沟通后,对患者样本进行全外显子测序(WES)。测序结果显示,患者CACNA1A基因第5号外显子存在新发突变c.674C>A(p.Pro225His)(见图5),其父母均未携带该突变。患者最终被诊断为以急性脑病为表现的偏瘫型偏头痛(HM),予每日口服氟桂利嗪 3 mg(0.2 mg/kg)预防发作。治疗 1.5 个月后随访复查头颅MRI显示,右侧半球皮层所有异常信号已完全消失(见图6)。因患者已完全恢复至发病前状态,且家属及患者对视频脑电图复查的依从性较低,未行该项随访检查。
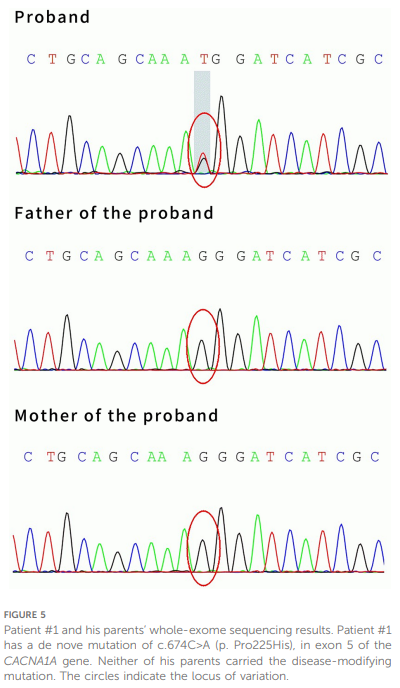
▲图5 病例1及其父母的全外显子组测序结果
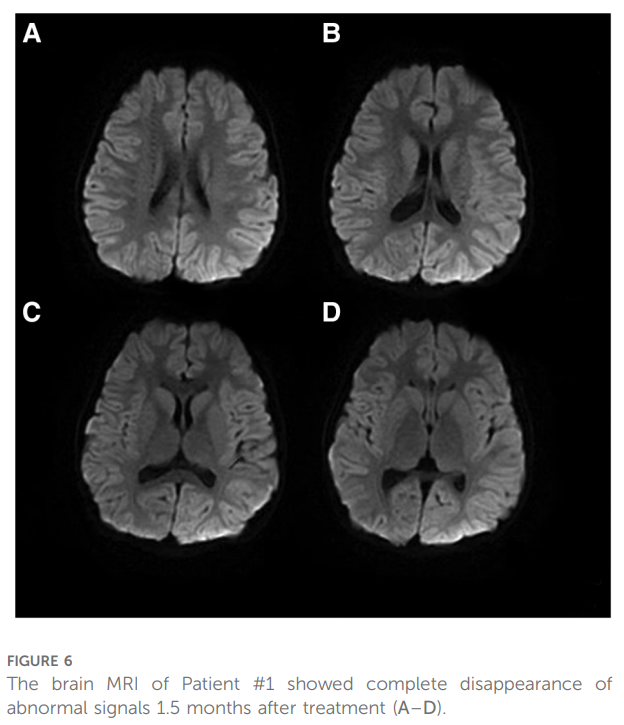
▲图6 病例1脑部MRI显示,治疗 1.5 个月后异常信号完全消失
病例2
患童男,5 岁,系非近亲婚配父母的次子,因局灶性发作伴嗜睡半天入住青岛大学附属医院。发作表现为双眼凝视、口唇发绀、上肢抖动,持续约1分钟,发病时体温38℃。血常规、血电解质、血氨、血糖、血钙均正常,头颅CT未见明显异常。神经系统查体:格拉斯哥昏迷评分(GCS)8分,四肢肌力、肌张力正常,双侧巴宾斯基征阳性,其余病理征阴性,脑膜刺激征阴性。患儿为足月顺产,既往智力及运动发育正常。1 岁时开始出现热性惊厥,至 2 岁时共发作 3 次;3 年前曾患病毒性脑炎;2 年前轻微头部外伤后出现头痛、恶心、呕吐,头颅CT检查正常,经休息后症状缓解。患儿母亲有癫痫发作史;姐姐被诊断为癫痫,目前予丙戊酸治疗,也曾有轻微头部外伤后发作及昏迷史。入院后立即完善相关检查:血生化、血同型半胱氨酸、血培养、单纯疱疹病毒检测均正常。因患儿不配合,入院时未行头颅MRI检查。结合家族史,建议患儿完善全外显子测序(WES),但家属拒绝。予甘露醇(2 g/kg/天)、甲泼尼龙(2 mg/kg/天)、美罗培南(60 mg/kg/天)、阿昔洛韦(27 mg/kg/天)及维生素B6(6 mg/kg/天)联合治疗。入院第2天,患儿再次发作,持续约 4 分钟,体温 38.2℃,予咪达唑仑处理后行腰椎穿刺术。脑脊液生化及细胞学检查:葡萄糖、氯化物、蛋白水平、细胞计数及分类均正常;脑脊液细菌、病毒、真菌检测均阴性。继续予冰毯、冰帽降温及上述联合治疗。入院第4天,患儿体温逐渐降至正常,未再出现发作,意识状态逐步好转。入院第13天行头颅MRI检查,结果正常;入院第 14 天,患儿恢复至发病前状态,同意完善全外显子测序后出院。结果显示ATP1A2基因第16号外显子存在错义突变c.2143G>A(p.Gly715Arg)(图7),该突变此前已有致病性报道。患儿母亲及姐姐均携带相同突变(图7)。经致病性预测工具分析:Mutation Taster判定该突变具有致病性;Polyphen 2预测其有害性得分为 1.000(敏感性:0.00;特异性:1.00);SIFT(截断值=0.05)预测其有害性得分为 0.000。
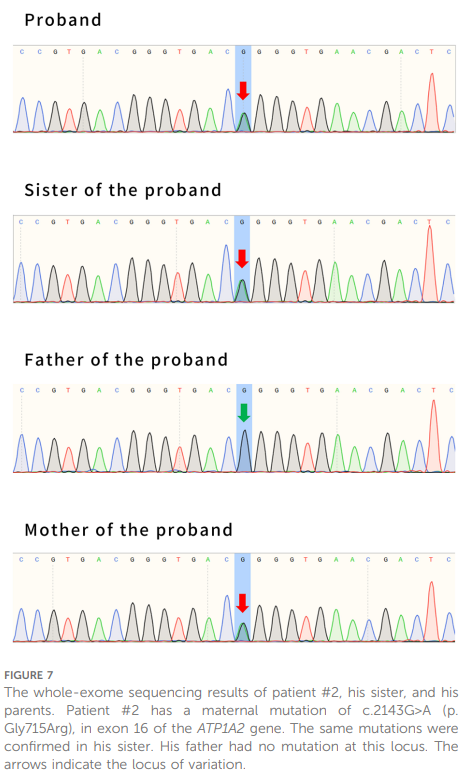
▲图7 病例2、其姐姐及其父母的全外显子组测序结果
经一系列检查后,该患者最终被诊断为偏瘫型偏头痛(HM)所致急性脑病,随后予氟桂利嗪 5 mg/日治疗。
讨 论
据估计,偏瘫型偏头痛(HM)的总体患病率为 0.01%。基于丹麦人群的研究数据,散发性偏瘫型偏头痛(SHM)患病率至少为 0.002%,家族性偏瘫型偏头痛(FHM)患病率至少为 0.003%。该病通常在 10 岁前或 20 岁前起病。HM发作的频率、强度和持续时间往往会随年龄增长而降低。情绪应激、剧烈体力活动、病毒感染和头部外伤是已报道的较为常见的HM发作触发因素。本文报道的 2 例患者均为男性,起病年龄均在 5 岁前。病例1的触发因素为轻度头部外伤;病例2的触发因素尚不明确,患儿在本次发病前出现流涕症状,因此研究人员推测可能与病毒感染有关。
HM可分为家族性(FHM)和散发性(SHM)两类。若一级或二级亲属中有类似症状者,则归类为FHM;反之则为SHM。根据致病基因突变的不同,FHM可进一步分为FHM1(OMIM #141500,由CACNA1A基因突变导致)、FHM2(OMIM #602481,由ATP1A2基因突变导致)和FHM3(OMIM #609634,由SCN1A基因突变导致)。SHM可由家族型致病基因的新发突变引起,也可由携带FHM致病突变但无症状的父母遗传而来。Bonemazzi等人的一项综述研究发现,与FHM患儿相比,儿童SHM患者的发作持续时间更长、病情更严重,尤其是在发病后的最初几年;而FHM患者的病程通常更长。
研究人员通过PubMed、Web of Science和Scopus数据库进行了全面检索,仅纳入报道了CACNA1A或ATP1A2基因突变患者完整临床信息的文献,最终筛选出 97 篇文献。结合本文的 2 例病例,共纳入 160 例患者(73 例携带CACNA1A基因突变,87 例携带ATP1A2基因突变),所有患者的临床特征总结于表1。
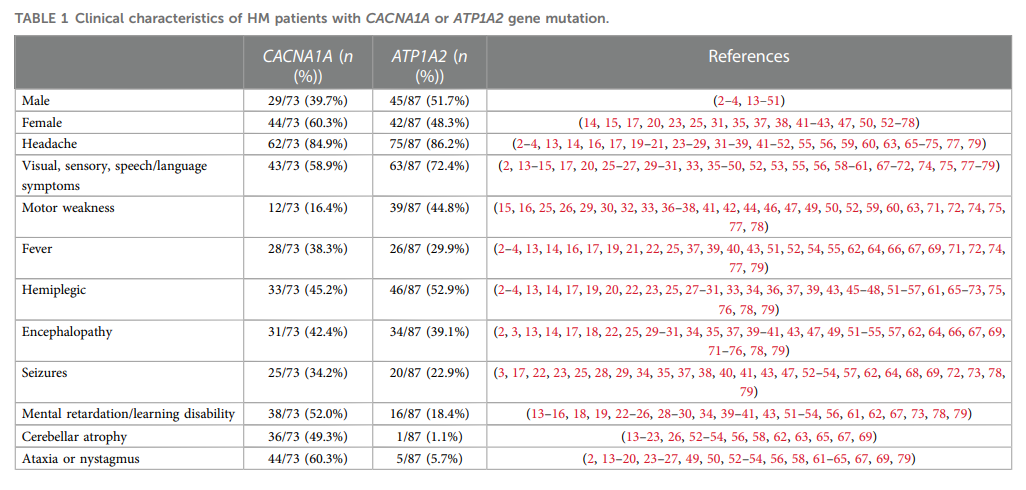
▲表1 CACNA1A或ATP1A2基因突变的HM患者的临床特征
研究人员发现,约半数患者表现为典型偏瘫型偏头痛(HM)发作,即出现完全可逆的视觉、感觉或言语症状,或运动无力。运动症状最常累及手、臂、足、腿、舌、面部及躯干。无力症状的进展呈渐进性,至少需5分钟,且症状通常单侧扩散,多数情况下表现为偏瘫。运动先兆的持续时间约为30分钟至24小时(平均 5 小时)。在本文报道的 2 例患者中,病例1的偏瘫症状持续超过 24 小时,与既往报道中对运动先兆症状的描述不符。本文的 2 例患者均未出现其他先兆症状(如感觉先兆、视觉先兆或失语先兆),这可能与患者年龄较小、表达能力不足有关。
然而,部分患者以脑病等非典型症状起病。在已报道的携带CACNA1A基因突变的HM患者中,42.4% 表现为脑病;在携带ATP1A2基因突变的患者中,39.1% 表现为脑病。脑病的具体表现为意识水平受损、发热或癫痫发作,且上述症状均持续超过 24 小时。轻度脑病患者仅表现为意识水平受损,而重度脑病患者可同时出现发热和癫痫发作。本文研究发现,所有伴发热的患者均存在脑病征象,且发热可能为中枢性发热。本组 2 例先证者均突发发热,无明确感染史,抗生素及退热药物治疗均无效,且均在体温恢复正常后临床表现开始好转。2 例患者均先出现局灶性发作,随后进展为全面性发作,其中病例1每次发作均以癫痫持续状态起病。病例1的全外显子测序结果显示,CACNA1A基因存在新发错义突变c.674C>A(p.Pro825His)。通过文献回顾,仅检索到 1 例Pro225His突变的相关报道:该患者为 1 例 8 岁早产女性,存在发育迟缓、言语及空间感知障碍、注意力不集中及癫痫病史,且至少经历过 4 次以头痛和发作为表现的HM发作。与之不同的是,病例1为足月出生,认知及运动发育正常,既往无癫痫发作史,本次同样表现为癫痫持续状态及偏瘫。但由于相关报道较少,目前尚无法明确该突变的临床表型。
病例2的全外显子测序结果显示,ATP1A2基因存在母系遗传的错义突变c.2143G>A(p.Gly715Arg)。通过文献回顾,目前已报道 3 例携带该突变的患者。表2总结了包括本病例在内的4例患者的临床特征:4 例患者均在儿童期起病,起病年龄为 1-6 岁,其中病例2的起病年龄最小;但因 4 例患者均以急性脑病起病,均出现了不同程度的诊断延迟。脑病急性期患者可表现为发热、嗜睡、偏瘫、癫痫发作或失语;除病例2急性期无头颅MRI数据外,其余 3 例患者在急性期均出现脑水肿。患者的脑病症状可能在数周内缓解,最长需 10 周恢复。结合这 4 例患者的临床表现,研究人员推测该突变可能与HM的早发及严重表型相关。
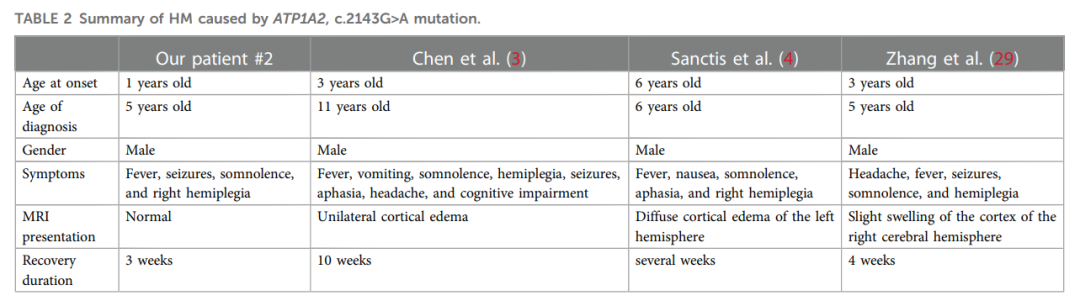
▲表2 由ATP1A2 c.2143G>A突变引起的HM总结
偏瘫型偏头痛(HM)合并急性脑病的发病机制目前仍不明确。Toldo等人报道了 1 例 8 岁女性散发性HM患者,其首次头颅MRI检查无异常,但神经影像学随访显示皮质肿胀进行性加重,弥散加权成像(DWI)呈轻度高信号,提示存在细胞内水肿。在患者偏瘫几乎完全恢复但失语仍持续时(发病第27天)行锝-99m-乙基半胱氨酸二聚体(99mTc-ECD)单光子发射计算机断层扫描(SPET),结果显示单侧大脑灌注显著减低。该发现提示存在原发性神经元功能障碍,“受损神经元细胞”对放射性药物的摄取减少。神经影像学表现可能与潜在的发病机制相关:离子通道功能障碍导致神经元去极化时间延长,进而引起水从细胞外转移至细胞内,最终导致细胞肿胀和神经元丢失。
大多数患者预后良好,但可能伴随神经系统异常及合并症。部分研究报道,高达60%的患者存在神经系统体征,最常见的异常包括小脑性共济失调、眼球震颤、姿势性震颤及动作笨拙。多数FHM1患者(通常超过半数家系)及少数FHM2患者可出现小脑体征,头颅MRI可见小脑萎缩。小脑萎缩可能随年龄增长逐渐显现。本文 2 例患者均经历多次HM发作,但均未出现小脑体征、小脑萎缩、运动发育迟缓或倒退,不过这可能与患者年龄较小有关,仍需长期随访。此外,Wada等人认为,CACNA1A基因突变本身而非昏迷发作频率,可能在进行性小脑萎缩的发病机制中起核心作用。FHM2患者出现小脑体征较为罕见,Spadaro等人报道的 3 例患者出现眼球震颤、步态共济失调、辨距不良及构音障碍等小脑症状,其中 1 例还存在小脑萎缩。有观点认为该表现可能与患者饮酒或使用镇静药物有关,但在其他家系中,未发现饮酒、药物、CACNA1A或脊髓小脑共济失调(SCA)基因突变、脑血管疾病等导致小脑体征的其他原因,提示这些体征可能是该家系FHM2表型的一部分。
研究人员发现,约半数携带CACNA1A基因突变的HM患者存在智力低下,这可能与该基因突变导致的钙通道功能受损有关。本组携带CACNA1A基因突变的病例1已发作 3 次HM,目前未发现智力低下或倒退表现,这可能与患者年龄较小或随访时间不足有关,未来需加强对该患者的观察。总体而言,HM合并急性脑病的潜在机制仍需更多研究探索。
目前,ATP1A2基因突变对HM患者认知功能的影响尚未得到详细评估。本文研究发现,ATP1A2基因对智力的影响多为一过性,HM症状缓解后可逐渐恢复至发病前水平。但Wang等人报道 1 例患者出现特定领域认知功能障碍,表现为数学能力及三维事物描绘能力受损,提示携带ATP1A2基因突变的HM患者可能出现永久性认知功能障碍。本文病例2的病史回顾显示,患者及其姐姐除计算能力较差外,其余方面均正常;患者母亲存在认知功能障碍,但拒绝接受智力测试。未来随访病例2时,认知功能检测是需重点关注的问题。
HM患者急性期仅需对症治疗即可缓解症状。部分报道指出,3 例携带CACNA1A基因突变的患者在脑病及脑水肿病程中联合使用类固醇和高渗溶液后,病情迅速恢复。对于FHM2患者,NMDA受体拮抗剂美金刚可预防谷氨酸能兴奋性毒性,艾地苯醌、dl-3-正丁基苯酞(dl-NBP)及传统抗偏头痛药物可保护线粒体功能。本文报道的 2 例以急性脑病起病的患者,急性期均接受对症治疗,予小剂量甲泼尼龙联合甘露醇治疗。由于病例2初期诊断未明确,未使用美金刚、艾地苯醌等药物。全外显子测序结果有助于临床未来选择更有效的治疗药物。
对于发作频繁、持续时间长或病情严重的患者,需进行预防性治疗。一项为期 11 年的儿童偏头痛单中心队列研究提示,氟桂利嗪应作为儿童HM患者的一线预防性用药。本组2例患者根据年龄、体重及药物耐受性,予氟桂利嗪 0.2 mg/kg/日,最大剂量不超过 5 mg/日。病例1和病例2分别接受氟桂利嗪治疗 8 个月和 2 个月,均耐受良好,未出现不良反应。目前随访期间,病例1偶有头痛,经休息可缓解,未再发生头部外伤及抽搐、脑病等严重发作,仍在继续随访中;病例2未再出现头痛或抽搐,这可能与随访时间较短有关,目前仍在继续随访。
急性脑病是儿童偏瘫型偏头痛(HM)严重发作的主要表现,这增加了诊断难度。对于表现为嗜睡、昏迷或抽搐且无结构性、癫痫性、感染性或炎症性病因的患者,医师在鉴别诊断时应考虑HM。疾病的早期识别与治疗有助于改善预后。全外显子测序可为诊断及分型提供依据。未来仍需开展多中心、大样本研究以探索该病的治疗策略。
参考文献:
Xiang Y, Li F, Song Z, Yi Z, Yang C, Xue J and Zhang Y (2023) Two pediatric patients with hemiplegic migraine presenting as acute encephalopathy: case reports and a literature review. Front. Pediatr. 11:1214837. doi: 10.3389/fped.2023.1214837

