Genome Biol | 华大团队及合作者发表新型高效染色质构象捕获方法,兼具可扩展性、成本效益及数据质量优势
时间:2026-05-05 17:16:03 热度:37.1℃ 作者:网络
染色质构象捕获技术是研究细胞中染色质三维结构的重要工具,可帮助认识和评估基因表达调控、DNA复制、重组和修复。传统Hi-C方法获得的是多种细胞的平均值,无法准确呈现不同细胞的异质性,且在分辨率和操作简便性方面存在不足。鉴于此,研究人员开发了更精确的单细胞Hi-C技术,但该技术在DNA损失、粘性末端连接效率仍有局限性。虽然有Dip-C、scCARE-seq等基于富集的替代方法,无需末端修复就能直接连接相邻片段,但可能会导致大量reads被丢弃。
近日,华大基因唐冲团队及国家卫健委男性生殖与遗传重点实验室团队合作在Genome Biology发表文章“Highly efficient chromatin conformation capture with post-enrichment in single cells by HiChew”,推出了新型高效染色质构象捕获方法HiChew。该方法结合了高效的黏性末端连接技术与基于PCR后甲基化的富集技术,能够处理大规模细胞数量以及对每个细胞进行超深度测序,更有效地检测染色质构象。研究显示,HiChew有效配对率约50%,并保持了高灵敏度。同时,HiChew的单细胞版本snHiChew实现了45%-50%的有效配对率,并能以5-10kb分辨率进行基因图谱绘制,基因组区间覆盖率达70%–80%。比较分析表明,HiChew在染色质区室、拓扑关联结构域(TAD)及环状结构的识别方面与传统Hi-C技术具有高度一致性。总之,HiChew方法在单细胞三维基因组结构分析中展现出迄今为止更高的效率,凭借可扩展性、成本效益及数据质量优势,HiChew为推进三维基因组结构研究提供了强大的平台。

HiChew开发与验证
HiChew技术的独特之处在于无需在扩增前进行预富集处理,其通过高效的黏性末端连接方法减少了扩增前所需的步骤。HiChew技术在PCR扩增前包含四个关键步骤:DNA链和蛋白质交联、DpnII酶切割GATC基序处的DNA、通过邻近连接技术将相邻酶切DNA末端连接、细胞裂解后,根据DNA量和实验需求选择基于接头连接的DNA文库构建方法(图1a)。扩增完成后,使用相应的甲基转移酶对连接位点进行m6A甲基化标记,并通过甲基化抗体对这些连接位点进行选择性富集测序。
HiChew采用DNA酶切与直接连接步骤,无需末端修复、生物素掺入、平末端连接及生物素富集等步骤。这种设计策略不仅提升了连接效率,PCR后富集还显著提升了染色质构象捕获效率,降低了细胞或DNA的潜在损失,且无需消耗大量测序资源。
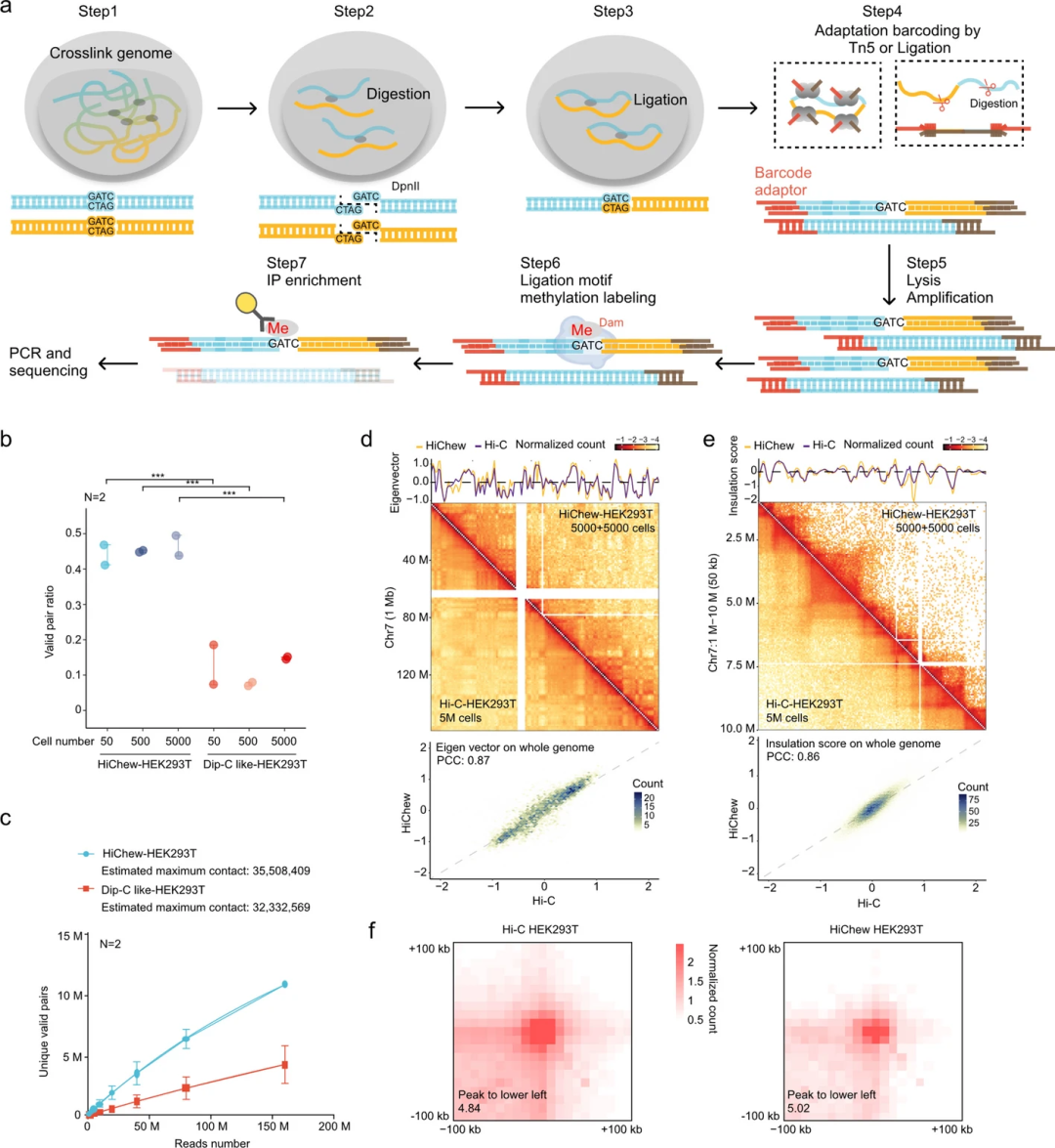
图1.HiChew方法操作流程及在染色质相互作用分析中的应用与验证。
为验证HiChew的性能,研究团队利用HEK293T细胞比较了无富集Dip-C和HiChew两种方法。评估显示,两种方法在顺式-反式比值和距离衰减曲线等性能指标上表现相近,无富集Dip-C显示出更高的未映射及无效配对。基准有效配对分析显示,HiChew实现了约50%的有效配对率(图1b)。虽然低于传统Hi-C方法的60%~90%,但显著优于无富集的Dip-C方法(8%-15%)。以上结果证明,HiChew在染色质构象捕获方面具有更优效率,产生的有效配对数约为无富集Dip-C的4倍。此外,通过GM12878细胞和组织的进一步验证,确认HiChew的有效配对比率稳定在40%-50%之间(图2d)。
与无富集Dip-C方法相比,HiChew产生的独特有效连接数量持续高出3-4倍。测序饱和度分析显示,HiChew仅使用50个细胞生成的最大连接数(3500万个)比使用相同细胞输入量的Dip-C方法所估算出的3200万个高10%(图1c),表明HiChew和Dip-C的理论最大文库复杂度相当。此外,在使用5000个细胞的情况下,HiChew在多个分辨率水平的表现与Hi-C相似。重要的是,HiChew方法具有极高的稳定性:在5000个、500个和50个细胞样本中,其批次间特征值相关系数分别达到0.96、0.98和0.95。总之,HiChew在保持灵敏度和准确性的同时提高了数据产量,为低输入量样本的染色质构象捕获分析提供了新的解决方案。
snHiChew开发与验证
进一步,研究团队通过PCR后富集技术对HiChew进行了优化,使其适用于单细胞染色质构象捕获,开发了创新的高通量、高效、单细胞核分析技术snHiChew。在交联处理后,经DNA酶切连接成串联体,随后利用HpyCH4V酶切DNA串联体,将细胞接种至96孔板中,完成dA尾端添加及第一轮带条形码接头的连接。在这一过程中,将收集的细胞核分离至次级培养板的不同组别中,通过PCR技术固定次级条形码(图2a)。对基因组文库进行PCR扩增后,利用甲基转移酶将连接处的损伤位点标记为m6A,利用m6A抗体富集有效的配对reads。值得注意的是,在设计条形码时需避开GATC基序,以避免被甲基转移酶靶向,从而防止数据出现偏差。
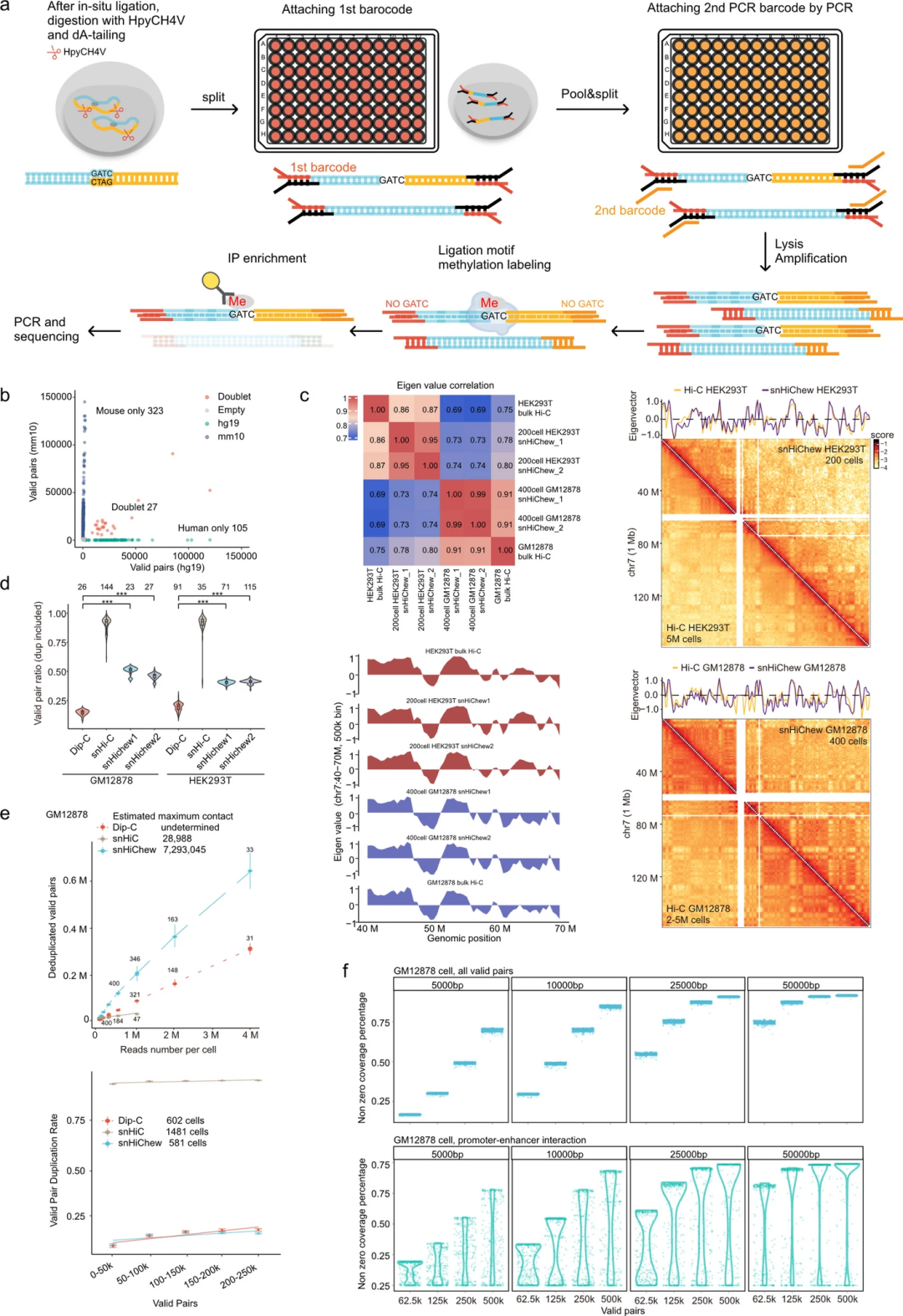
图2.snHiChew方法的开发与验证
随后,研究团队使用HEK293T与NIH/3T3细胞混合样本评估了snHiChew的准确性和效率。在首批400个细胞中,snHiChew检测到27个双细胞群,碰撞率为0.059(图2b)。后续1,000个细胞样本经双细胞群过滤后,碰撞率保持在0.062。UMAP聚类分析显示,两种细胞类型实现了清晰区分。
在snHiChew与无富集Dip-C、snHi-C的比较中,三种方法显示出相似的顺式-反式比值和距离衰减曲线。与批量样本类似,伪批量snHiChew在关键参数AB区室结构和TAD绝缘性上高度复现了批量Hi-C的结果(图2c)。在更高分辨率下,snHiChew与Dip-C(LiMCA)在环状结构信号富集方面表现出高度一致性,且snHiChew的类别分布更接近金标准Hi-C的结果。以上结果表明,snHiChew不仅能有效区分单细胞,其染色质构象捕获精度也与成熟的批量方法相当。
在HEK293T细胞中,Dip-C和snHi-C获得的有效配对率分别约为12%–20%和70%–90%,snHiChew约为45%(图2d)。在GM12878细胞中,snHiChew的有效配对率达到约50%,比Dip-C高出4倍,比snHi-C低,但snHi-C具有最高的重复率(图2d-e)。在不同测序深度下去重后,snHiChew为7,293,045,显著高于snHi-C的28,988,表明其在保留信息性有效配对方面具有更高的捕获灵敏度(图2e)。当每个细胞拥有50万个有效配对时,snHiChew在10kb基因组区间的覆盖度超过80%,在5kb基因组区间约为70%(图2f)。与reads数相当的其他技术相比,snHiChew展现出更优的覆盖性能,能够在单细胞中识别出更多增强子-启动子相互作用。
snHiChew的应用分析
在HEK293T细胞分析中,snHiChew获得了约1178个有效细胞,每个细胞平均包含25万-200万个独特的有效配对。(图3)分析确定了位于B区室内的若干长编码基因,发现了一些活跃的组蛋白修饰,例如CNTNAP2启动子上的H3K4me3信号以及基因体上的H3K27ac修饰。研究进一步探讨了B区室中染色质结构与基因激活之间的关系,发现TAD绝缘性降低与更高的基因表达水平及染色质可及性相关。同时,研究还发现熔解状态与凝聚状态之间在染色质定位上的差异(图3g-h)。以上结果表明,染色质结构、可及性与基因活性之间存在关联,有助于完善关于异染色质相关区室内转录过程的模型。
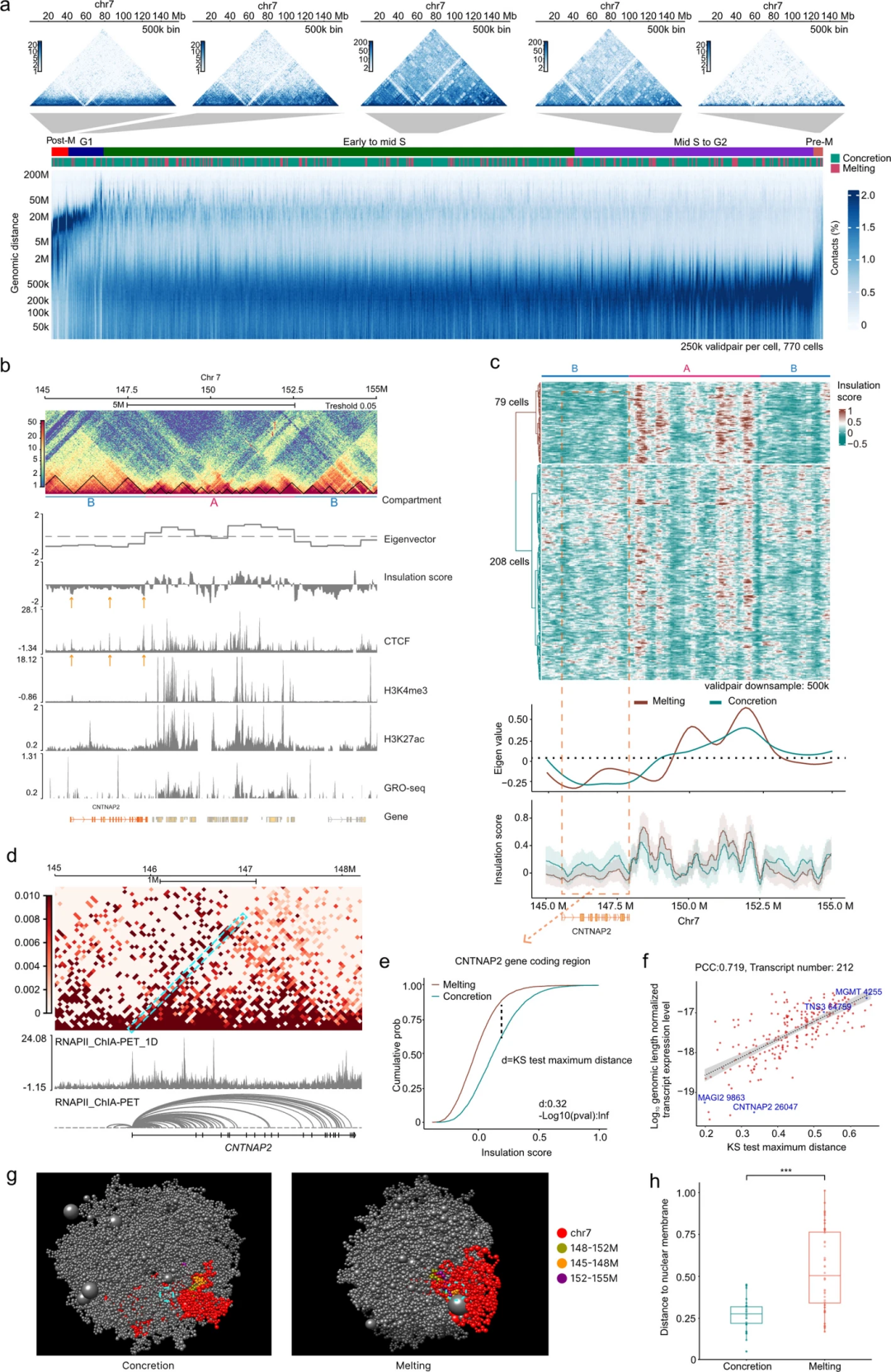
图3.转录活动期间染色质结构的分析。
此外,研究证实CTCF介导的染色质绝缘减弱与基因表达活性增强之间存在显著相关性。CTCF下调会增强染色质解聚和基因表达,转录抑制会降低染色质解聚程度。这些结果凸显了染色质结构、转录与基因调控之间的复杂关系。
结语
该研究开发的新型染色质构象捕获技术HiChew和snHiChew,有效解决了该领域的一个主要问题:生物素富集在单细胞分析中的整合效率低下,导致大量有效相互作用数据丢失。研究显示,HiChew和snHiChew在不造成DNA损失的前提下,有效配对比率可达约50%,较传统无富集方法提升6倍,显著优化了资源利用效率和数据质量。snHiChew在保持45%–50%效率的同时,实现了5kb-10kb分辨率的高精度图谱绘制,且区间覆盖率达70%-80%。
总之,HiChew在保持灵敏度和准确性的同时,降低了测序成本、提高了数据产出量,且无需大幅增加测序深度,为基因组学和表观遗传学研究提供了成本效益更高的解决方案,将成为单细胞三维基因组研究的革命性工具。
原文信息:
Chen, Z., Xie, Y., Zhang, C. et al. Highly efficient chromatin conformation capture with post-enrichment in single cells by HiChew. Genome Biol 27, 127 (2026). https://doi.org/10.1186/s13059-026-04059-1

