上海交大Science子刊:揭示剪接体突变通过谱系特异性RAS激活驱动黑色素瘤发生
时间:2026-05-05 13:05:24 热度:37.1℃ 作者:网络
背景介绍
RNA剪接因子突变是人类癌症中最常见的突变类型之一,其中SF3B1是突变频率最高的剪接因子。值得注意的是,SF3B1的突变热点呈现明显的谱系特异性:K700E突变主要发生于血液系统恶性肿瘤(如骨髓增生异常综合征和慢性淋巴细胞白血病),而R625突变则特异性富集于黑色素瘤(包括葡萄膜黑色素瘤、黏膜黑色素瘤和皮肤黑色素瘤),突变频率高达14-29%。然而,这种癌症类型特异性的分子机制长期未明。为何SF3B1 R625突变偏好发生在黑色素细胞中,而K700E却几乎不见于黑色素瘤,一直是肿瘤生物学中一个引人入胜的谜题。
研究思路
针对这一难题,上海交通大学医学院附属第九人民医院/上海精准医学研究院的曾汉林研究员与北京基因组研究所刘肇祺研究员团队联合多家单位,通过系统分析黑色素瘤和血液肿瘤患者的RNA测序数据,结合正常黑素细胞和K562白血病细胞中引入SF3B1不同突变(R625H/K700E)的等基因细胞模型,揭示了两种突变在异常剪接上的根本差异。他们发现,SF3B1 R625H突变比K700E突变诱导了更强的隐蔽3'剪接位点(cryptic 3'ss)使用。机制上,隐蔽分支点(BPS)上游和周围富集的多聚腺嘌呤(poly-A)序列为R625H突变提供了多个替代分支点腺嘌呤,使其能够更有效地识别并激活隐蔽剪接位点。这一剪接偏倚导致RAS抑制因子NF1优先发生错剪接并经由无义介导的mRNA降解(NMD)而沉默,从而引起RAS过度激活,加速黑色素瘤进展。该研究建立了从“序列偏好→基因选择性错剪接→癌症特异性”的完整逻辑链,为理解SF3B1热点突变的谱系特异性提供了全新机制框架。相关内容以Spliceosomal mutation drives melanoma tumorigenesis via lineage-specific RAS activation为题,发表在Science Advances!

图片解析
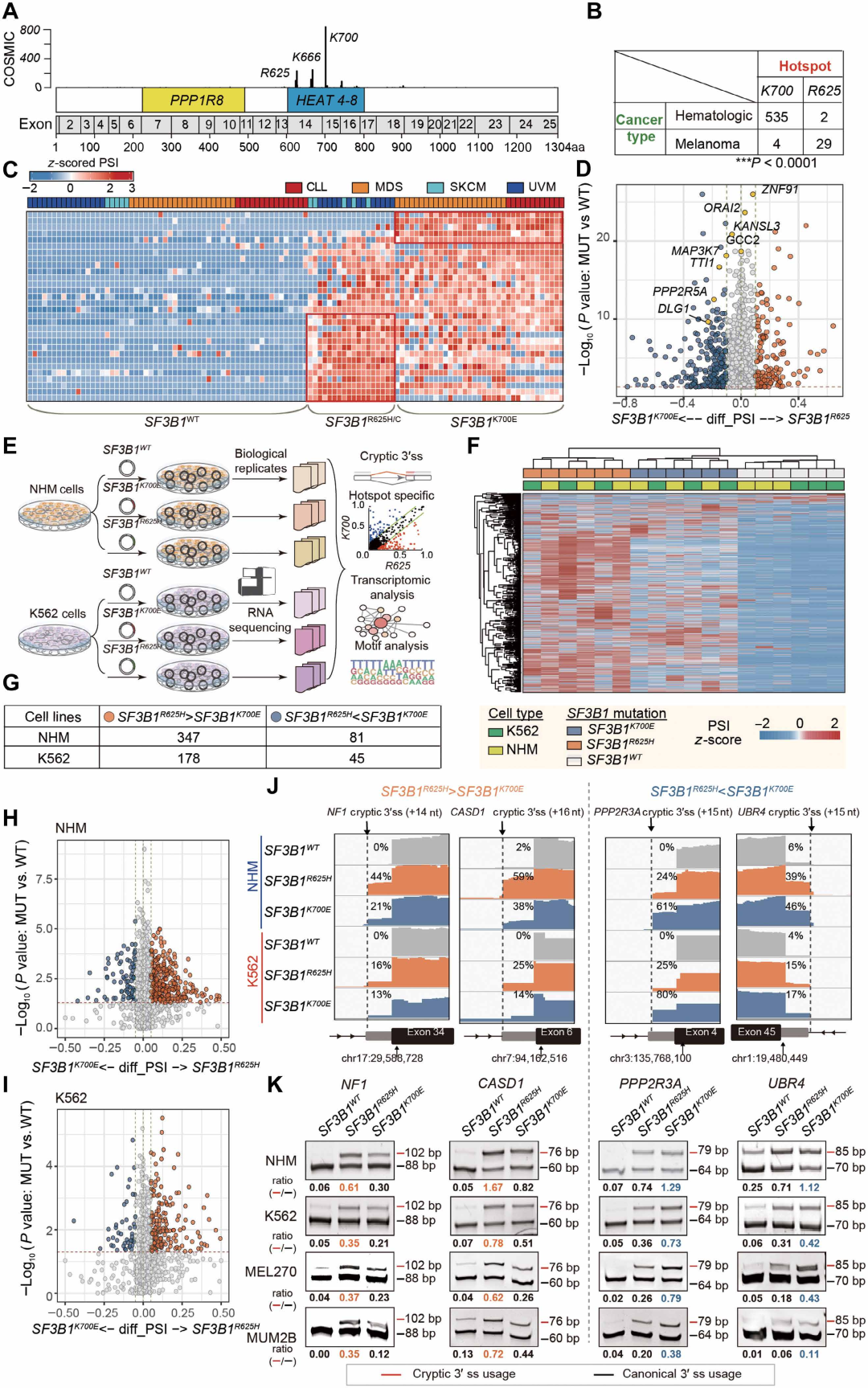
图1. SF3B1 R625突变与K700E突变产生不同的错剪接谱: (A) 基于COSMIC数据库的SF3B1突变图谱,K700、K666和R625是最常见的突变残基。(B) 列联表显示血液肿瘤与黑色素瘤患者中SF3B1突变类型分布,R625与黑色素瘤显著相关(Fisher精确检验,P<0.0001)。(C) 患者样本中前30个隐蔽3'ss事件的PSI值层次聚类热图,R625与K700突变样本呈现不同的剪接模式。(D) 泛癌患者样本中K700突变与R625突变诱导的隐蔽3'ss变化火山图,先前报道的MAP3K7、PPP2R5A、DLG1等事件在两种热点间差异使用。(E) 等基因细胞模型构建示意图:在黑素细胞(NHM)和K562细胞中分别引入SF3B1 WT、R625H和K700E。(F) 等基因细胞中隐蔽3'ss事件的PSI热图,R625H与K700E突变细胞同样呈现不同剪接模式。(G) 列联表显示NHM和K562细胞中优先与R625H或K700E相关的隐蔽3'ss事件数量,R625H偏好事件显著更多。(H-I) NHM(H)和K562(I)细胞中R625H相对于K700E诱导剪接变化的火山图。(J) IGV图展示代表性基因(R625H偏好:CASD1、NF1;K700E偏好:PPP2R3A)的隐蔽3'ss使用差异。(K) RT-PCR验证上述基因的剪接差异。
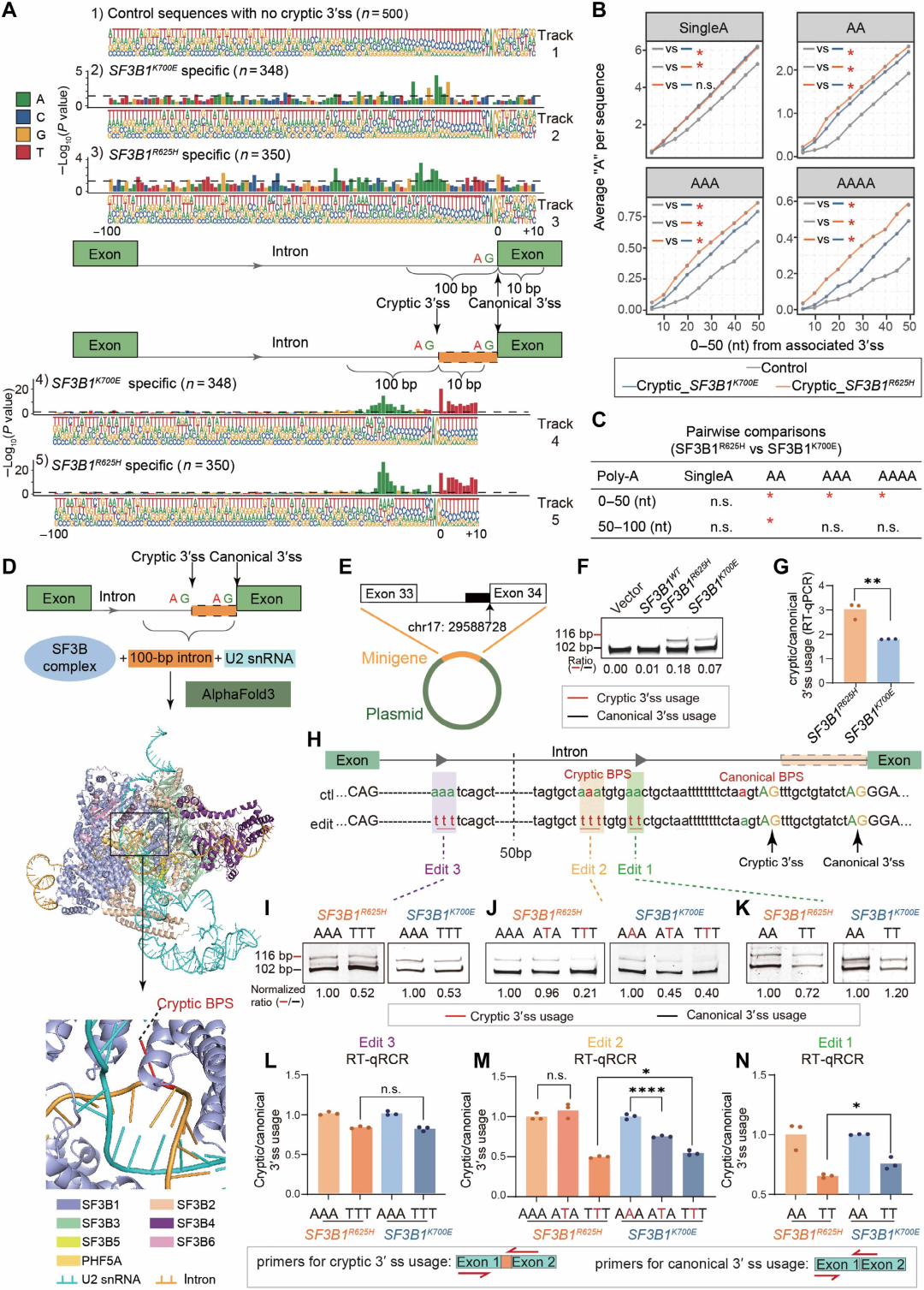
图2. 隐蔽AG位点上游多聚腺嘌呤(poly-A)富集导致SF3B1 R625H更强的隐蔽3'ss活性: (A) 对照序列(无隐蔽3'ss)、K700E偏好隐蔽3'ss序列、R625H偏好隐蔽3'ss序列中隐蔽AG位点上游100 bp至下游10 bp的核苷酸组成。R625H组中poly-A(连续A)序列显著富集。(B) 各组中隐蔽AG位点上游100 bp内连续A的出现频率量化,R625H组AA、AAA和AAAA频率显著高于K700E组。(C) 隐蔽AG位点上游0-50 bp内连续A的出现频率,R625H组富集更明显。(D) AlphaFold3预测的SF3b复合物与U2 snRNA及内含子序列的复合物结构,显示31 nt上游的隐蔽分支点腺嘌呤嵌入AAA序列中。(E) NF1第33-34外显子minigene构建示意图及关键碱基编辑位点。(F-G) 不同SF3B1突变细胞中minigene剪接产物的凝胶电泳(F)和定量(G),R625H诱导更高的隐蔽剪接比例。(H-N) 不同编辑位点(edit1:分支点上游AA→TT;edit2:AAA→TTT;edit3:远端poly-A突变)对隐蔽剪接的影响。edit2(分支点AAA全部突变)完全消除R625H的隐蔽剪接能力,edit1(上游AA突变)对R625H的抑制更强,edit3无显著影响。
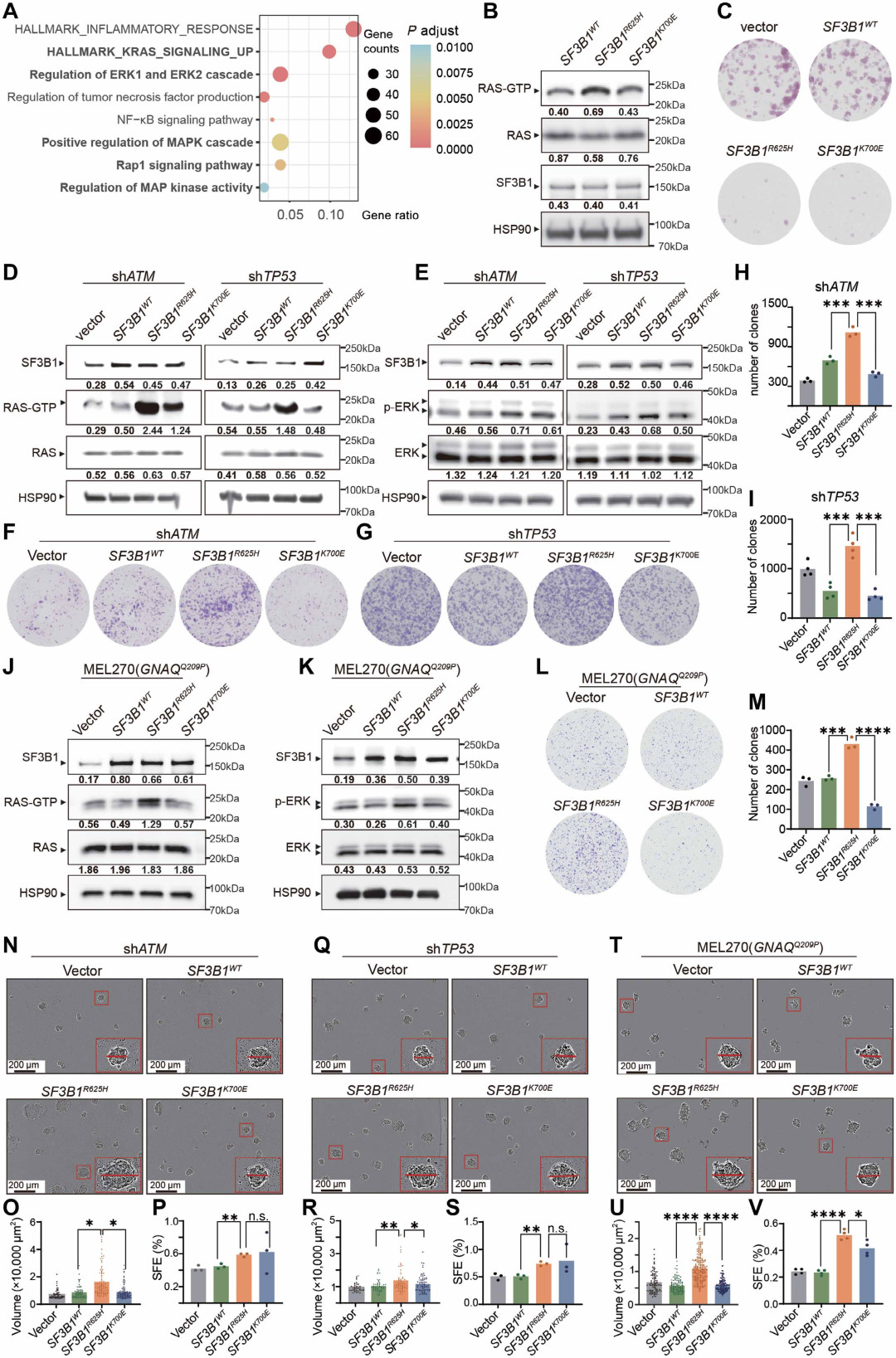
图3. SF3B1 R625H突变比K700E突变介导更强的RAS激活: (A) TCGA黑色素瘤队列中SF3B1突变与野生型样本的差异表达基因通路富集分析,显示炎症反应和RAS相关通路(RAF-MEK-ERK)显著富集。(B) MUM2B黑色素瘤细胞中不同SF3B1突变过表达后的RAS活性(RAS-GTP水平)检测,R625H组RAS活性更强。(C) 菌落形成实验显示直接过表达突变SF3B1抑制细胞生长,提示需要额外遗传改变耐受剪接应激。(D) ATM或TP53敲低背景下,R625H细胞表现出更强的RAS激活。(E) ATM/TP53敲低后,R625H细胞中ERK磷酸化(p-ERK)水平更高。(F-I) 菌落形成实验及定量:ATM(F,H)或TP53(G,I)敲低下,R625H细胞菌落形成能力显著强于K700E。(J-M) 在携带GNAQ Q209P突变的MEL270细胞中,R625H同样诱导更强的RAS激活(J)、ERK磷酸化(K)和菌落形成(L-M)。(N-V) 肿瘤球形成实验:ATM(N-P)或TP53(Q-S)敲低下MUM2B细胞以及MEL270细胞(T-V)中,R625H细胞形成的肿瘤球体积更大、形成效率更高。
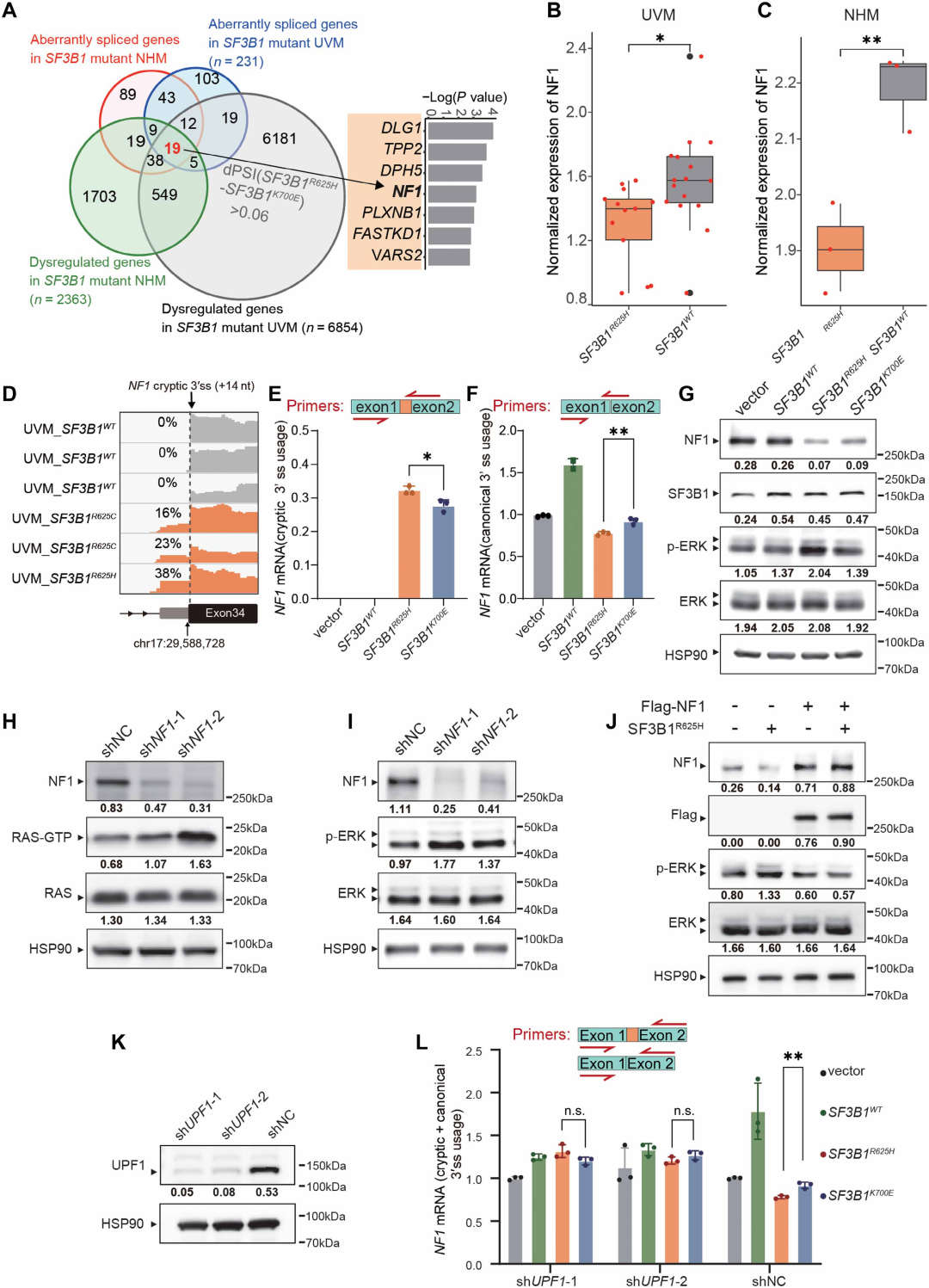
图4. SF3B1 R625H通过NF1的异常剪接促进RAS激活: (A) TCGA UVM肿瘤和NHM细胞中SF3B1突变与野生型之间差异表达和差异剪接基因的Venn图,重叠基因包括NF1等。(B-C) UVM患者(B)和NHM细胞(C)中NF1 mRNA表达水平,R625H突变样本中NF1表达显著降低。(D) IGV图显示UVM患者样本中NF1基因座的隐蔽3'ss使用,R625H突变样本中隐蔽读段明显。(E) RT-qPCR显示R625H细胞中异常剪接的NF1 mRNA水平显著高于K700E。(F) R625H细胞中正常剪接的NF1 mRNA水平显著降低。(G) Western blot:R625H细胞中NF1蛋白水平下降,p-ERK升高。(H-I) NF1敲低(H)和过表达(I)实验证实NF1是RAS和MAPK通路激活的关键介导者。(J) R625H细胞中异常剪接的NF1转录本引入提前终止密码子,触发NMD。(K) UPF1敲低验证。(L) UPF1敲低后,R625H细胞中总NF1 mRNA水平恢复,证明NMD是NF1下调的主要机制。
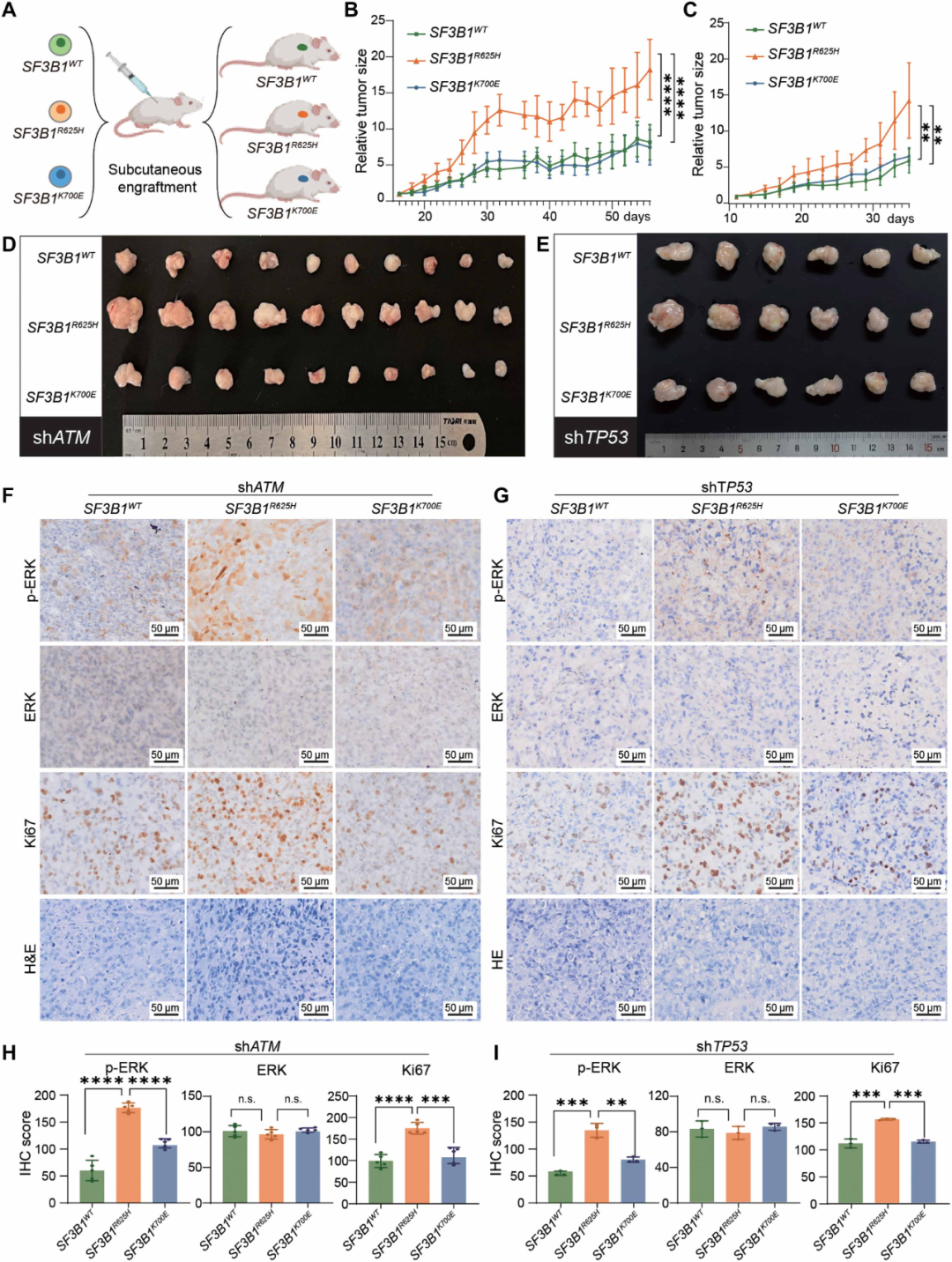
图5. SF3B1 R625H突变通过RAS激活驱动体内黑色素瘤进展: (A) 小鼠皮下成瘤模型构建示意图。(B) ATM敲低背景下各SF3B1突变细胞的肿瘤体积变化曲线,R625H组肿瘤生长最快(n=10,P<0.0001)。(C) TP53敲低背景下肿瘤体积变化曲线(n=6,P<0.01)。(D-E) ATM(D)和TP53(E)敲低背景下各组肿瘤代表性照片。(F-G) 肿瘤组织的免疫组化染色,检测p-ERK、总ERK和Ki67。R625H组p-ERK和Ki67阳性率最高。(H-I) 免疫组化评分的定量分析,R625H组显著高于K700E组和WT组。
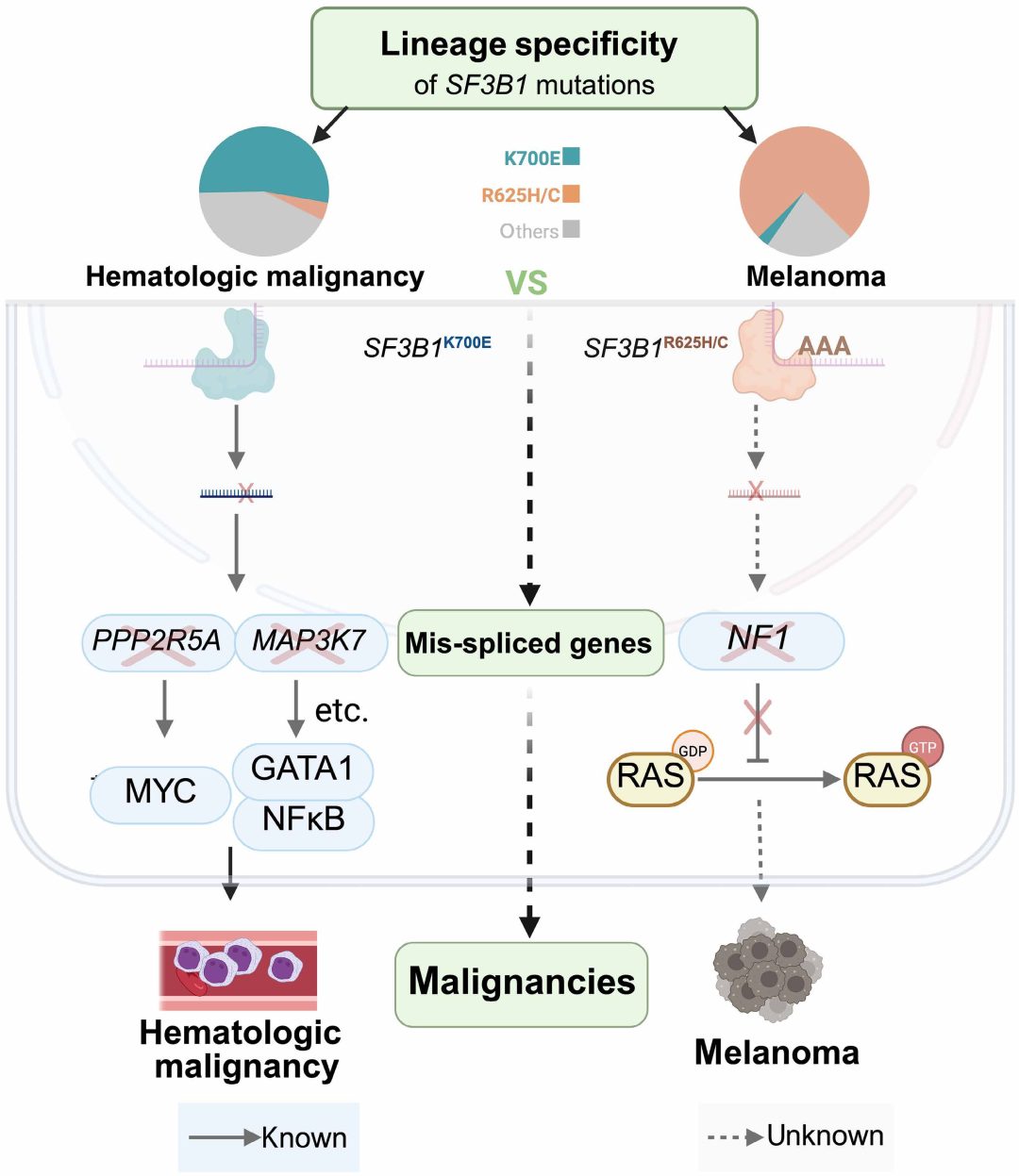
图6. SF3B1 R625突变驱动黑色素瘤发生的机制模型: SF3B1 R625通过优先诱导NF1前体mRNA的错剪接,导致NF1蛋白缺失,进而激活RAS信号通路,最终促进黑色素瘤的发生发展。
结论
本研究首次系统揭示了SF3B1不同热点突变(R625H vs K700E)在剪接产物上的功能差异并非源于细胞类型差异,而是由其固有的序列识别偏好所决定。R625H突变能够更有效地利用隐蔽分支点周围的多聚腺嘌呤序列作为替代分支点,从而更强力地诱导多种基因(尤其是NF1)的隐蔽3'ss使用和NMD降解。NF1的缺失导致RAS信号通路过度激活,最终驱动黑色素瘤的发生和进展。该工作不仅为SF3B1 R625突变在黑色素瘤中的谱系特异性提供了机制性解释,也为靶向RAS-MEK通路的治疗策略提供了理论依据,并为其他剪接因子突变相关肿瘤的研究提供了新范式。
原文链接:
https://www.science.org/doi/10.1126/sciadv.adz8289

