病例报告|复发性结外型颅内外沟通性Rosai-Dorfman病1例
时间:2025-02-05 12:10:08 热度:37.1℃ 作者:网络
摘 要 报告1例复发性结外型颅内外沟通性Rosai-Dorfman病。患者,中年女性,2017年5月以“头皮肿物术后10月,复发1月”入院,入院诊断为“颅内外沟通性占位性病变”,全麻下显微手术切除肿物并行颅骨成形术,术中将肿物和累及的硬脑膜和颅骨全切除,病理诊断为“Rosai-Dorfman病(Rosai-Dorfman disease, RDD)”,全身未发现淋巴结肿大,术后口服强的松1月。之后定期复查,未见复发。2022年12月右额皮肤出现一个结节样肿物,活检病理诊断为RDD,口服沙利度胺无效,手术切除。2023年10月MRI发现右额顶叶交界区窦镰旁肿物,再次手术切除,病理诊断为RDD,随访8个月无复发。提示累及中枢神经系统的RDD可以多次复发,需长期随访;显微手术切除颅内外沟通性RDD安全有效,复发者可以再次手术。
关键词
Rosai-Dorfman病;中枢神经系统;复发;颅骨;淋巴结
Rosai-Dorfman病(Rosai-Dorfman disease, RDD),即窦组织细胞增生伴巨淋巴结病(sinus histiocytosis with massive lymphadenopathy, SHML),发病率约1/20万 [1],常表现为颈部淋巴结无痛性肿大,25%~43%的患者有淋巴结外器官受累。仅累及淋巴结外器官而无淋巴结肿大的占20%,称为结外型RDD,其中,累及中枢神经系统(central nervous system, CNS)者罕见,占结外型的不足5%[2],颅内外沟通性RDD更罕见。本组收治1例结外型颅内外沟通性RDD,报告如下。
1 临床资料
患者,女,汉族,56岁。2017年5月以“头皮肿物术后10月,复发1月”入院。患者于2016年7月无明显诱因出现头痛,持续性,头顶部为主,无恶心、呕吐或抽搐,未就医。1个月后,头顶出现一个约1 cm×1 cm水泡状肿物,破溃后头痛较前缓解,之后,破溃部位长出一肉芽样肿物,逐渐增大至核桃大小,在当地医院手术切除,但未做病理检查,术后切口愈合不佳,反复换药后愈合。2个月后MRI发现“右顶部中线旁长T1长T2占位性病变,自皮下至硬膜下,局部硬膜增厚,颅骨似有缺损”。2017年4月原手术部位再次出现一个直径约1 cm的灰黄色肿物,偶有分泌物,不疼,遂来我院就诊,入院诊断为“颅内外沟通性占位性病变(脑膜瘤可能性大;嗜酸性肉芽肿不除外);颅骨缺损(右顶部)”。查体见右顶部中线旁头皮淡红色肿物,无压痛及破溃,余无异常。颅骨切线位X线见“颅顶结节状软组织突起,约5 mm×7 mm,界限清晰,顶骨骨质不连续,可见范围约7 mm局限性缺损”(图1)。MRI见“右顶骨部分缺损,局部可见结节状软组织影,T1WI、T2WI均呈等信号,T2FLAIR呈等-稍低信号,形态不规则,边界欠清,增强扫描呈明显均匀强化,2.1 cm×2.0 cm,病灶深面脑膜局部增厚,邻近脑组织受压,其内可见斑片状稍长T1稍长T2信号区域。MRV可见病灶与上矢状窦分界不清,上矢状窦腔内未见充盈缺损”(图1)。彩超报告“双侧颈部及锁骨上窝未见明显增大的淋巴结”。全麻下行颅内外沟通瘤切除并颅骨成形术(图2),术中见头皮肿物与颅内相连,中线及右顶骨中线旁各一骨质缺损,缺损处可见肿物自颅内长出,呈灰褐色,质脆,血供一般,帽状腱膜被侵袭,围绕颅骨缺损及肿物周围钻孔,内板骨质酥软,切除异常骨质至骨质正常,见肿物穿破硬膜,周围硬脑膜增厚,进一步扩大骨窗至显露正常硬脑膜,马蹄形剪开硬膜,翻向中线,肿物呈灰红色,质韧,血供丰富,与上矢状窦壁粘连,压迫脑表面,与脑表面界限不清,显微镜下分离粘连,全切肿物,可见脑表面被侵袭,切除受累的硬脑膜,硬脑膜缺损和颅骨缺损分别用自体骨膜和钛网修补。整个肿物连同受累的硬脑膜、骨质和头皮标本送组织病理学检查。病理诊断为“皮肤、纤维结缔组织及骨小梁间大量浆细胞及淋巴细胞浸润,其间可见较多组织细胞,局灶炎性肉芽肿组织及脓肿形成,免疫组化结果显示S-100(+)、CD138(大量浆细胞阳性)、CD38染色提示病灶中还有大量浆细胞;CD1a、Langerin及CD56(-);CD79α及MUM1部分细胞(+);Kappa及Lambda未见限制性表达;Ki-67增殖细胞指数约5%,支持RDD伴重度慢性炎”。骨髓检查显示“增生性骨髓、巨核细胞易见、粒红可见病态造血现象、网状细胞易见”。术后恢复顺利,切口愈合良好,口服强的松1个月。每年定期复查颅脑MR平扫+增强扫描,未见复发。
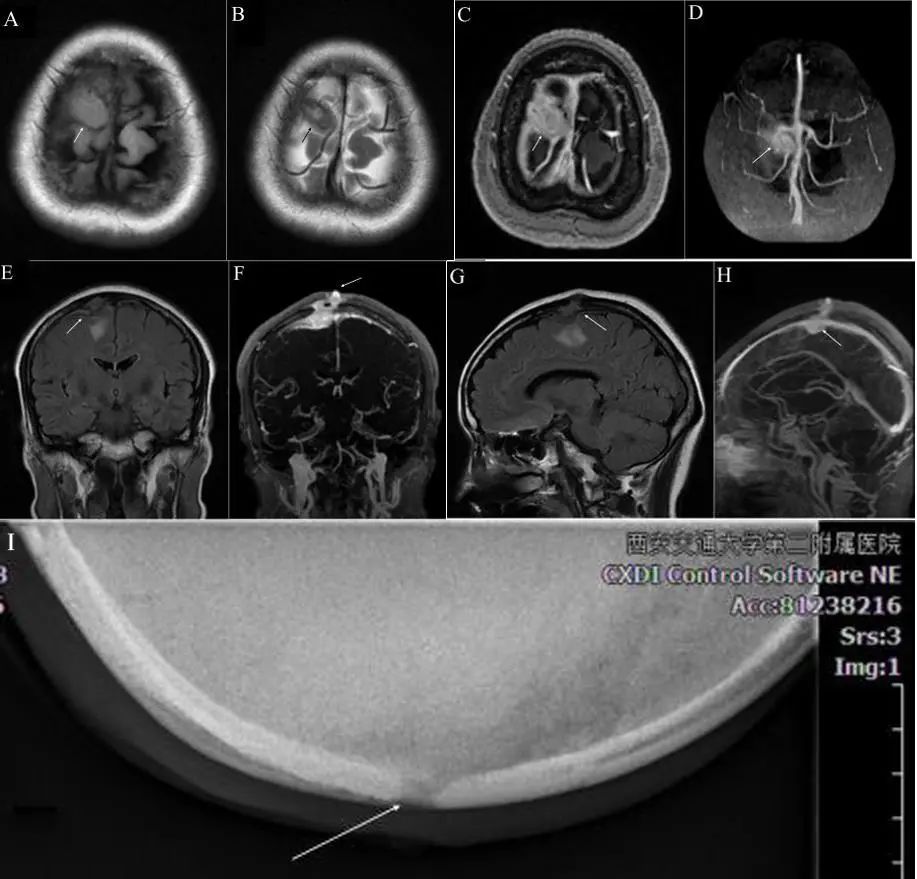
图1 首次术前影像学 A.轴位T1WI;B.轴位T2WI;C.轴位T1FLAIR增强;D.轴位MRV可见病灶与上矢状窦关系密切;E.冠状位T2FLAIR;F.冠状位MRV;G.矢状位T2FLAIR;H.矢状位MRV;I.颅骨切线位X线。Fig.1 The images before the first craniotomy
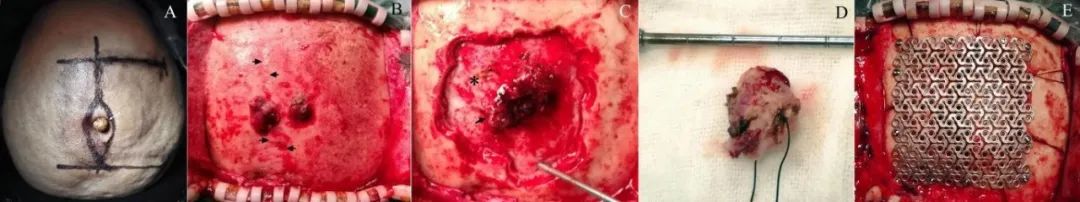
图2 首次术中所见 A.外观;B.肿物穿破颅骨;C.肿物侵袭硬脑膜;D.肿物标本;E.钛网修补颅骨缺损。Fig.2 The observations during the first craniotomy
2022年12月右额皮肤出现一个结节样肿物,肉红色,逐渐增大,2023年3月在我院穿刺活检诊断为RDD,口服沙利度胺,但肿物仍进行性增大,2023年6月再次入院。查体见右额皮肤有一直径约3 cm肉红色肿物(图3),界清,表面凹凸不平,无压痛或触痛,可随头皮活动,右颊部皮肤可见肉红色斑。局麻下行“右额皮肤病损切除术”,术中见皮下肿物质地坚韧,血供丰富,边界清晰,周围骨质无异常,完整切除。术后恢复顺利,切口愈合良好,病理报告“慢性炎伴感染,淋巴细胞增生及组织细胞增生,部分区域淋巴组织生长活跃”。
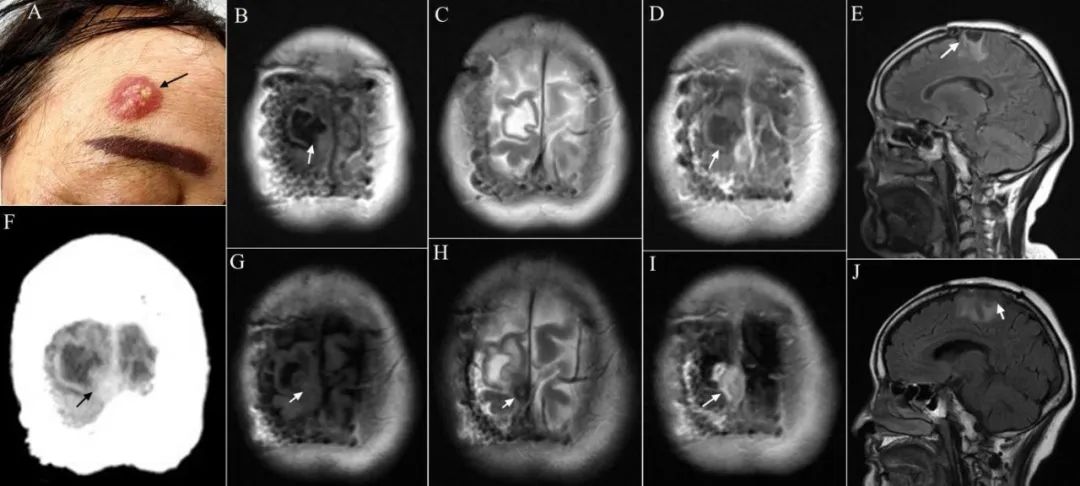
图3 随访过程中影像学表现 A~E, 2023年4月;F~J, 2023年10月。A.右额皮肤病灶;B.轴位T1WI;C.轴位T2WI;D.轴位T1增强扫描;E.矢状位T1WI;F.轴位CT;G.轴位T1WI;H.轴位T2WI;I.轴位T1增强扫描;J.矢状位T1WI。Fig.3 The images during the follow-up
2023年10月无明确原因出现左侧肢体抽搐,持续几十秒,发作时意识清楚,无其他不适。复查MRI报告“右侧顶骨缺损,新增右额顶叶交界区中线旁异常信号影,T1WI呈稍低信号,T2WI呈稍高信号,FLAIR呈稍低信号,周围可见高信号,增强扫描呈显著强化”(图3)。遂以“颅内RDD术后6年,间断抽搐发作10余日”之主诉入院,查体:神志清楚,左下肢肌力5-级,左侧Babinski征可疑阳性,余(-)。超声报告“双侧颈部及锁骨上窝、双侧腹股沟区及腹膜后均未见增大淋巴结”。ECT报告“右顶骨局部术后改变,颅骨弥漫性骨质代谢增高;胸腰椎骨质代谢改变;全身其余骨质代谢未见明显异常”。自身免疫抗体检查提示ANA抗体阳性。全麻下行右顶部开颅窦镰旁肿物切除术(图4),术中见原骨窗缘骨质异常,灰色鱼肉样组织自原钛网中央网孔挤出,拆除钛网,硬脑膜显著增厚,中央被灰红色组织突破,切除异常骨质,骨质酥软易出血,马蹄形剪开硬脑膜,分离粘连,见肿物基底位于上矢状窦壁和大脑镰,主体位于纵裂内被脑组织遮盖,有包膜,与脑表面粘连紧密,肿物质地不均匀,大部分呈灰白色,质地坚韧,有钙化,中央小部分呈灰红色,质地稍软,血供不甚丰富,肿物嵌入大脑半球内侧面,周围脑组织软化,近全切除肿物,仅残留上矢状窦侧壁少许瘤基底,电凝之。切除异常硬脑膜,人工硬膜修补缺损,保留颅骨缺损。术后恢复顺利,切口愈合良好,口服抗癫痫药物。病理诊断为RDD,Bcl-6(+),Bcl-2少数(+),P53(+)1%,Ki-67增殖细胞指数约20%。2024年6月复查MR未见病变复发(图4),再次入院在全麻下行PEEK材料颅骨修补术,术后恢复顺利,继续口服抗癫痫药物,定期随访。
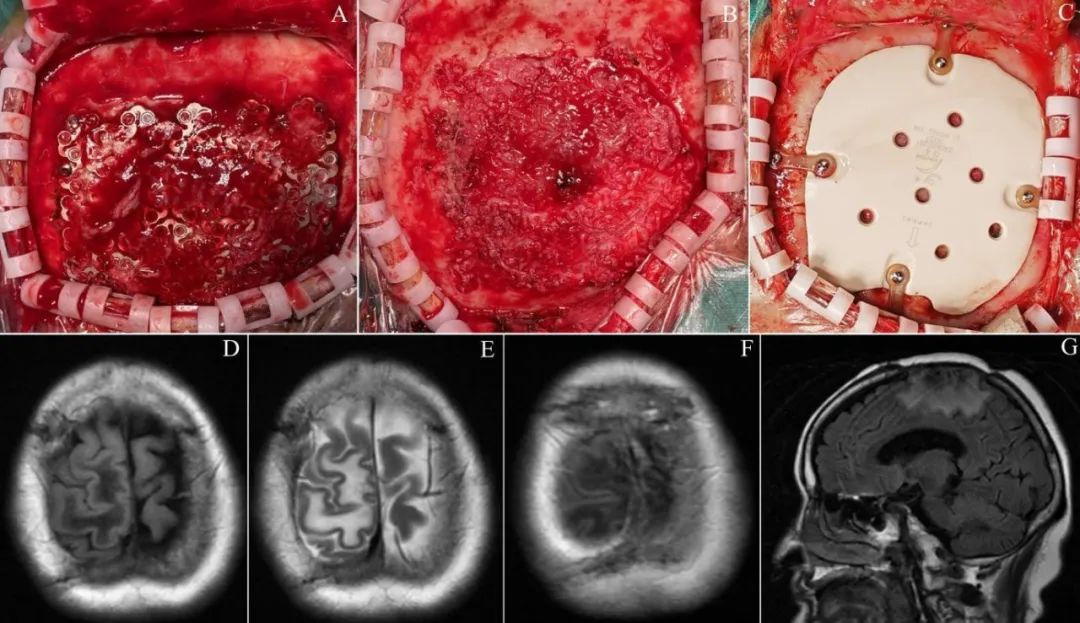
图4 复发病灶术中所见以及颅骨修补术 A.病变自钛网突出;B.病灶侵袭硬脑膜;C.8个月后PEEK修补颅骨缺损;D.2024年6月轴位T1WI;E.轴位T2WI;F.轴位T1增强扫描;G.矢状位T1WI。Fig.4 The recurrent lesion observed during the surgical procedure and the cranioplasty
2 讨论
RDD病是一种罕见的非朗格汉斯细胞组织细胞增生症,以往认为是一种良性特发性非肿瘤性疾病,但近年发现30%~50%的RDD病例有MAPK/ERK通路突变,如 KRAS、NRAS、MAP2K1、ARAF、PTPN11等,提示有一部分RDD是肿瘤性疾病,2022年第五版《WHO造血与淋巴组织肿瘤分类》将其归入树突细胞和组织细胞肿瘤 [3]。
RDD的临床和影像学表现皆缺乏特异性,早期诊断困难。影像学检查首选MR,累及硬脑膜者需与脑膜瘤鉴别。本病例MR显示T1WI、T2WI均呈等信号,T2FLAIR呈等-稍低信号、周围水肿严重、局部颅骨溶骨性破坏,增强扫描呈均匀强化、有脑膜尾征,与文献基本一致,但不足以据此与脑膜瘤鉴别。有报道[4]认为RDD呈T2FLAIR低信号有助于将其与脑膜瘤鉴别。本例T2FLAIR呈稍低信号,似乎支持这一点。研究[4-5]发现RDD的ASL呈低灌注,MRS脂质峰和N-乙酰天冬氨酸峰升高,有助于鉴别诊断。由于本例初诊为脑膜瘤或嗜酸性肉芽肿,未进行ASL和MRS检查,这是不足之处。对于疑似累及重要血管的病变,术前应完成MRA和MRV。本例颅骨破坏严重,尚需与朗格汉斯细胞组织细胞增生症(颅骨嗜酸性肉芽肿)、转移瘤、淋巴瘤鉴别。
确诊RDD依靠组织病理学和免疫组化染色。镜下结外型RDD缺乏特征性,免疫表型对诊断帮助更大[2]。RDD免疫组化S-100、CD68、CDl63、OCT2、cyclin D1阳性[3]、EMA、 GFAP、CDla阴性。本例S-100(+)、CD138(+)、CD68(+)、GFAP(-)、CD1a(-)、Langerin(-)、CD56(-),符合RDD特点。
RDD曾被认为是一种自限性疾病,绝大多数预后良好,但资料显示死亡率可达7%~12% [1, 6],累及CNS者少有自发缓解。据报道[7],RDD完全或部分缓解后复发率可达16.3%。本病例多次复发,且复发过程中Ki-67增殖指数呈增高趋势,提示其可能并非都是良性自限性,需长期随访。
RDD尚无统一的治疗方法,对于有症状或造成脑局部受压的颅内或颅内外沟通性RDD,手术切除仍是合理的选择,但术后常需联合其他治疗,例如免疫调节剂、糖皮质激素、化疗、靶向药物、放疗等 [8]。对于单发颅内RDD,全切除可长期缓解。由于放疗对RDD效果有限,本次手术全切除后未予放疗,应用强的松1个月,获得6年余的缓解。复发RDD可以再次手术切除。若病变累及重要结构,不必勉强追求全切除。
综上所述,累及CNS的RDD可以多次复发,需长期随访;显微手术切除颅内外沟通性RDD是安全有效的,复发者可以再次手术。
参考文献:
1. ABLA O, JACOBSEN E, PICARSIC J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease[J]. Blood, 2018, 131(26): 2877-2890.
2. PARKHI M, CHATTERJEE D, KASHYAP D, et al. Primary Rosai-Dorfman disease of the central nervous system: A clinical, histological, and molecular appraisa[J]l. Neuropathology, 2024, 44(5): 366-375.
3. RAVINDRAN A, RECH K L. How I Diagnose Rosai-Dorfman Disease[J]. Am J Clin Pathol, 2023, 160(1): 1-10.
4. TYAGI G, KONAR SK, MEHTA S, et al. Management of intracranial Rosai-Dorfman disease: An institutional experience[J]. J Clin Neurosci, 2024, 127:110758.
5. 刘升阳, 姚玉鑫, 赵丽涛, 等. 颅内原发性Rosai-Dorfman病伴上矢状窦后段血栓一例并文献复习[J]. 北京医学, 2022, 44(5): 432-435, 440.
6. BRUCE-BRAND C, SCHBEIDER J W, SCHUBERT P. Rosai-Dorfman disease: An overview[J]. J Clin Pathol, 2020, 73(11): 697-705.
7. NASANY R A, REINER A S, FRANCIS J H, et al. Rosai-Dorfman-Destombes disease of the nervous system: A systematic literature review[J]. Orphanet J Rare Dis, 2022, 17(1): 92.
8. 刘婷,曹欣欣. Rosai-Dorfman病的诊治进展[J]. 基础医学与临床, 2022, 42(11): 1785-1790.
【引用格式】周毕盛,权瑜,王举波,等. 复发性结外型颅内外沟通性Rosai-Dorfman病1例[J]. 中国神经精神疾病杂志,2024,50(12):746-749.
【Cite this article】ZHOU B S,QUAN Y,WANG J B,et al.A case report of recurrent extranodal extra- and intracranial communicating Rosai-Dorfman disease after surgery[J]. Chin J Nervous Mental Dis,2024,50(12):746-749.
DOI:10.3969/j.issn.1002-0152.2024.12.007

