罕见卵巢「两性母细胞瘤」检出DICER1致病突变,NGS助力少见肿瘤临床诊疗
时间:2025-02-13 12:20:32 热度:37.1℃ 作者:网络
本文描述了一名 16 岁女性,主诉腹部肿块延伸至剑突。腹部和盆腔CECT显示右侧卵巢肿块大,呈囊实性,甲胎蛋白升高。患者接受了右侧输卵管卵巢切除术。冰冻切片组织病理学显示性索间质肿瘤。考虑到患者年龄较小,进行了保留生育能力的分期手术。最终的组织病理学和免疫组织化学显示为两性母细胞瘤[中分化至低分化Sertoli‑Leydig细胞成分(80%)+幼年型粒层细胞瘤(JGCT,20%)]。最终FIGO分期为IA期,G3。患者接受了辅助化疗(4 个周期的BEP)。NGS分析显示DICER1基因发生突变。患者治疗后 12 个月随访无复发,血清肿瘤标志物AFP在正常范围内。
背 景
两性母细胞瘤(gynandroblastoma)是一种罕见的卵巢性索间质肿瘤,通常表现为两种不同组织学的混合,即粒层细胞瘤和Sertoli‑Leydig细胞瘤(SLCT)分化。传统上,两性母细胞瘤大多具有成人型粒层细胞,但具有幼年粒层细胞瘤成分的两性母细胞瘤极为罕见。甲胎蛋白(AFP)是卵巢生殖细胞肿瘤公认的肿瘤标志物,它在SLCT中也与一些特殊的组织学标准有关。Sertoli‑Leydig细胞瘤和两性母细胞瘤通常与DICER1突变有关。DICER1突变是一种罕见的基因突变,使个体易患许多其它良性和恶性肿瘤。因此,本文介绍了针对这种罕见病例(AFP升高且存在DICER1突变的两性母细胞瘤)的治疗经验,因为这种罕见病例没有标准化的治疗方案指南。
TIPS|两性母细胞瘤
近年来,随着分子生物学的进展,发现了许多肿瘤携带某种特定的基因异常改变,可以协助明确肿瘤类别。第5版WHO分类显示,超过 90% 的成人型粒层细胞瘤(adult granulosa cell tumour,AGCT)含有FOXL2基因突变,在分别为 60% 和 30% 的幼年型粒层细胞瘤(juvenile granulosa cell tumour,JGCT)中可以检测到AKT1和GNAS基因的激活改变。Sertoli‑Leydig细胞瘤是一类向睾丸方向分化的卵巢性索‑间质肿瘤,又称为男性母细胞瘤(arrhenoblastoma)或睾丸母细胞瘤(androblastoma),该肿瘤可分为 3 个不同的分子亚型,①DICER1基因突变型:年轻女性,肿瘤呈中~低分化,可以伴有网状或异源性成分;②FOXL2基因突变型:绝经后女性,肿瘤呈中~低分化,不伴有网状或异源性成分;③DICER1或FOXL2基因野生型:中年女性,肿瘤多呈高分化,不伴有网状或异源性成分。微囊性间质瘤含有CTNNB1基因或低频APC基因突变,并且可能是家族性腺瘤性息肉病的结肠外表现。
此外,第5版WHO分类重新引入了两性母细胞瘤(gonadoblastoma),该肿瘤包含女性成分(包括AGCT或JGCT)和男性成分(包括Sertoli细胞瘤或Sertoli‑Leydig细胞瘤),最常见的组合形式是含有较多的Sertoli‑Leydig细胞瘤成分和较少的JGCT成分。免疫组化法染色显示,两种肿瘤成分通常对性索来源组织标志物(如抑制素和FOXL2)呈阳性表达。大多数两性母细胞瘤是良性的,罕见复发。

▲摘自《第5版WHO女性生殖器官肿瘤分类的更新及解读》
病 例
患者女,16 岁,主诉腹胀加重以及过去 6 个月月经不调而就诊。体格检查发现腹部肿块向头部延伸几乎抵至剑突。腹部和盆腔CECT显示右侧卵巢肿块较大,呈囊实性。患者的肿瘤标志物CA-125、B-HCG和LDH均在正常范围内,但AFP水平升高为 263 ng/ml(正常值 0-40 ng/ml)。患者接受了右侧输卵管卵巢切除术。冰冻切片组织病理学(HPE)报告显示性索间质肿瘤(SCST)。此外,考虑到患者年龄较小,还进行了保留生育能力的完全分期手术。该病例的最终FIGO分期为IA期,G3。大体检查显示卵巢肿块大小为 25×20 cm,外表面光滑。切面显示实性和囊性区域(图1)。最终HPE显示该病例为非常罕见的混合性索间质肿瘤(两性母细胞瘤)[SLCT(80%)+幼年型颗粒细胞瘤(20%)]。Sertoli‑Leydig细胞成分中等至低分化,存在少量散在的间质细胞成分,以及局灶性网状特征。有丝分裂明显(15-20 个有丝分裂/HPF)。幼年型成分显示存在多个有分泌物的实性结节(类似于格拉夫卵泡,图2)。免疫组织化学(IHC)标志物抑制素、钙结合蛋白、波形蛋白、细胞角蛋白和Melan A./Mart 1为阳性,而IHC标志物EMA、CK7和SALL4为阴性。通过下一代测序(NGS)技术发现患者携带致病性DICER1突变。建议患者进一步行与DICER1综合征相关的家族性基因筛查和咨询。肿瘤医生建议基于不良预后因素(如Sertoli‑Leydig细胞成分分化中等至较差、肿瘤大小 >30 cm和 15-20/10 HPF有丝分裂)进行辅助化疗(4 个周期的BEP)。治疗后 12 个月的随访中,患者无复发,血清肿瘤标志物AFP在正常范围内。
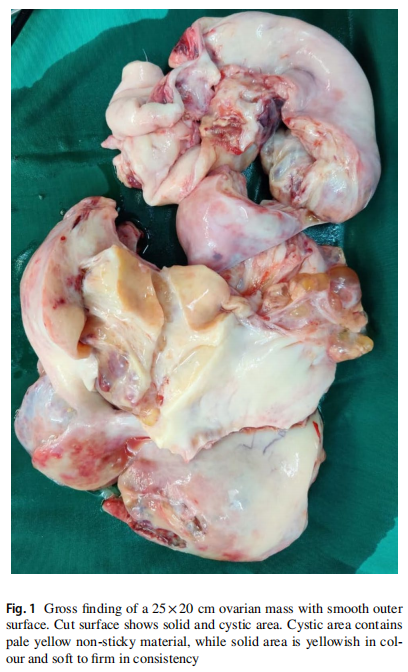
▲图1 大体检查发现一个 25×20 cm的卵巢肿块,外表面光滑
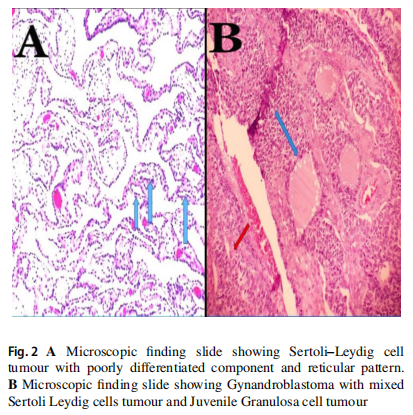
▲图2
A.显微镜下发现Sertoli‑Leydig细胞肿瘤,分化较差,呈网状结构;B.显微镜下发现两性母细胞瘤,混合有Sertoli‑Leydig细胞肿瘤和幼年型粒层细胞瘤
讨 论
两性母细胞瘤的诊断主要取决于组织学特征。两性母细胞瘤的先决诊断标准要求次要肿瘤成分(GCT中的Sertoli‑Leydig细胞或SLCT中的粒层细胞)应占整个肿瘤的至少 10%。因此,足够的肿瘤取样对于准确诊断至关重要。尽管大多数报告的GCT病例属于成人型,但也发现了幼年型。GCT的成人型和幼年型细胞的组织学相同;因此,诊断这些肿瘤并不困难。一项研究发现,尽管存在几种有助于鉴别诊断的免疫组织化学标记,但这些标记可普遍用于诊断每种卵巢性索间质肿瘤,并且它们并非两性母细胞瘤所特有的。代表性的性索基质细胞标志物包括抑制素、钙结合蛋白、SF1、WT-1、CD99、细胞角蛋白和波形蛋白。MART-1/melan-A是SLT和类固醇细胞瘤特有的标志物。在本文病例中,JGCT和SLT成分均对抑制素、钙结合蛋白、细胞角蛋白和波形蛋白呈阳性。
2020 年世界卫生组织指南根据分化程度和特点,将卵巢SLCT分为高分化、中分化、低分化、异质性和网状肿瘤。卵巢SLCT的预后与临床分期和病理分级相一致。临床分期I-II期患者 5 年癌症相关生存率为 100%,晚期肿瘤患者为 0%;按病理分级划分的 5 年癌症相关生存率分别为 100%、96% 和 33%。文献中的一项研究表明,SLCT复发预后较差,主要影响初次诊断后的年轻患者。由于两性母细胞瘤发病率极低,因此很难描述其生物学行为。根据报告的病例,该病似乎呈良性病程。
SLCT的治疗建议取决于生育期望,因为保留生育能力的手术可以在完整的手术分期下进行。由于复发风险较高,中分化或低分化病例通常建议术后辅助化疗。常用的化疗方案是博来霉素、依托泊苷、顺铂和紫杉醇联合方案。最有效的化疗方案尚无明确的共识。本文研究遵循了相同的治疗指南。
甲胎蛋白(AFP)是卵巢生殖细胞肿瘤的显著肿瘤标志物,同时在具有某些特殊组织学模式和胃肠道异源性成分的SLCT中也发现了该标志物。由于它在原发性和复发性肿瘤中均有分泌,因此它是一种重要的血清学随访标志物,同时可指示疾病对治疗的反应。在本文研究中,发现AFP水平在手术后恢复正常,并在随访中保持在正常范围内。
大约 50–60% 的SLCT发生在DICER1胚系突变携带者中。医学文献中的研究得出结论,中分化和低分化SLCT成分经常共存,并且几乎所有病例都与DICER1突变有关。在患有DICER1综合征的个体中,SLCT是最常见的性索间质卵巢肿瘤。有家族史的SLCT患者通常有甲状腺异常,例如甲状腺肿和甲状腺瘤。这些融合发现表明携带DICER1突变的个体有家族遗传史。绝大多数DICER1综合征癌症具有局限于肿瘤组织的双等位基因DICER1突变,这可能代表局限于肿瘤的突变。医学文献中的研究表明,突变分析研究应结合DICER1胚系突变检测和肿瘤组织检测。这种方法通常可以检测出几乎所有与DICER1突变相关的SLCT。
Hughes等人强调,卵巢SCLT大多为单侧,但也有报道出现双侧肿瘤,但两个卵巢肿瘤内不同的DICER1体系热点突变证明它们是相互独立的肿瘤。这些发现支持以下观点:双侧卵巢SLCT确实是独立事件,并不一定代表复发或疾病转移。基于这些原因,Hughes等人对该病例进行长期随访,因为与复发性肿瘤相比,异时性SLCT的预后更好。
参考文献:
Limbachiya, D., Tiwari, R., Kumari, R. et al. Case of Gynandroblastoma of the Ovary with Raised AFP and Associated DICER 1 Mutation. J Obstet Gynecol India (2024). https://doi.org/10.1007/s13224-024-02005-4


