汇总!肺动脉高压遗传学之通道病基因分析
时间:2022-03-05 11:21:53 热度:37.1℃ 作者:网络
肺动脉高压 (PAH) 是一种罕见的、进行性且通常致命的疾病,其特征是肺动脉发生明显变化,导致肺血管阻力增加。PAH 的估计患病率为 4.8-8.1 例/百万儿童发病和 5.6-25 例/百万成人发病。失调的血管、炎症和免疫细胞有助于增加血管阻力和肺动脉和小动脉的增殖性重塑。这些病理变化增加了心脏右心室的负荷,最初导致右心室肥大,后来导致死亡率高的右心衰竭。
PAH 可能由遗传、表观遗传和环境因素以及基因与环境相互作用引起,其中遗传对疾病风险的贡献因环境暴露而改变。家族中可遗传的 PAH (HPAH) 的早期遗传学研究将骨形态发生蛋白受体 2 型 ( BMPR2 ) 确定为主要致病基因。来自患者登记处和大型 PAH 队列的遗传数据表明,约 70% 的家族病例是由罕见的有害BMPR2变异引起的。然而,家族性病例仅占所有 PAH 病例的一小部分。BMPR2变异体和 BMPR2 信号通路中的其他基因(ACVRL1、ENG、SMAD9和GDF2) 是约 12-20% 的散发性特发性 PAH (IPAH) 病例的原因,而与其他疾病 (APAH) 或药物/毒素相关的 PAH 很少见。最近,外显子组和基因组测序研究发现了 BMPR2 通路之外的新 PAH 风险基因,包括三个通道基因:ATP 结合盒亚家族成员 8 ( ABCC8 )、ATPase 13A3 ( ATP13A3 ) 和钾双孔结构域通道 ( KCNK3 )。一项研究报告了第四个通道基因水通道蛋白 1 ( AQP1 ) 但尚未被复制。在个别病例中确定 PAH 的遗传原因可能对临床管理产生影响,包括治疗(单模式与多模式治疗)、手术干预和移植决策,以及相关疾病的筛查。至少 13% 的成人发病和 43% 的儿童发病 PAH 病例可以由遗传原因解释 。
表 1. 多项研究中的 PAH 因果通道病基因和相关变异等位基因频率/特征。

ATP 结合盒亚家族成员 8 ( ABCC8 )
在 TGFβ/BMP 通路之外,通道病基因ABCC8是 PAH 最常见的原因之一,约占病例的 1%。ABCC8是 ATP 结合盒 (ABC) 转运蛋白基因超家族的成员,编码磺酰脲受体 1 (SUR1) 蛋白,一种 ATP 敏感的钾通道调节亚基。钾通道在维持细胞静息膜电位和细胞内钙浓度方面发挥重要作用。ABCC8在胰岛细胞中高度表达,其作用是释放胰岛素,隐性突变导致先天性高胰岛素血症和新生儿糖尿病。ABCC8通过对一组 99 例儿科和 134 例成人发病的 PAH 病例进行外显子组序列分析,首次将其鉴定为 PAH 风险基因。在一名 10 岁的 IPAH 患者中发现了一种罕见的、预测的有害的从头错义变异。从头变异很少见,当有害时,可能与早期的高死亡率有关,因此不会传播给下一代。因此,对整个 PAH 队列进行了ABCC8基因变异筛查,并在 7 名患有 IPAH、HPAH 或 APAH-先天性心脏病 (APAH-CHD) 的不相关患者中发现了罕见或新的错义变异。与 PAH 的常染色体显性遗传一致,所有个体都是罕见的ABCC8变体的杂合子。ABCC8 _c.718G>A;p.240A>T 变体在一个家族中与 PAH 分离。在英国 (UK) PAH 队列中进行的一项复制研究确定了四个额外的变体、三个错义变体和一个剪接变体。一个ABCC8变异杂合子也有一个罕见的TBX4移码突变;其他携带者都没有任何已知的风险基因突变。在美国/英国合并中,大约一半的患者患有成人发病和一半的儿童发病。与两个独立的欧洲人口对照组(每个n > 33,000 人)相比,欧洲 PAH 病例中ABCC8变体的统计富集分析表明ABCC8的富集率是 3 倍PAH 病例中的变异,在病例和对照之间预测的良性同义变异的频率没有显著差异。
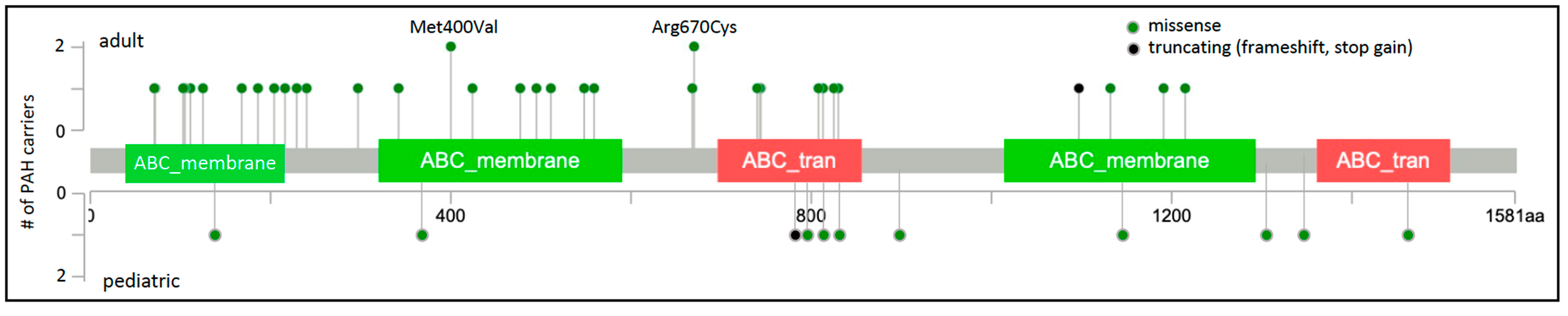
图 1. ABCC8二维蛋白质示意图。
ATPase 13A3 ( ATP13A3 )
TP13A3编码一种跨膜阳离子转运蛋白,最近显示该转运蛋白可转运多胺。多胺是正常细胞生长和增殖所需的小代谢物,据报道,在多种癌症和最近的 PAH中血浆浓度升高。ATP13A3的遗传变异首先在英国 NIHR 生物资源稀有疾病研究的 1083 例 PAH 病例的基因组测序研究中得到报道。在排除了 7 个已确定的 PAH 因果基因变异的病例后,与对照组相比,在病例中观察到更高频率的蛋白质截断变异。还观察到蛋白质截断加错义变体的趋势,但未达到全基因组显性。确定了 10 个具有独特罕见、预测有害变体的不相关 IPAH 病例:三个移码变体、两个停止增益变体、一个剪接变体和四个错义变体。全球多项研究证实了PAH中ATP13A3的独立验证。在 331 例不相关的 IPAH 病例的中国队列中发现了另外四种错义变异。在上述欧洲儿科 PAH 队列中,在 APAH-CHD 病例中发现了一种新的错义变异。
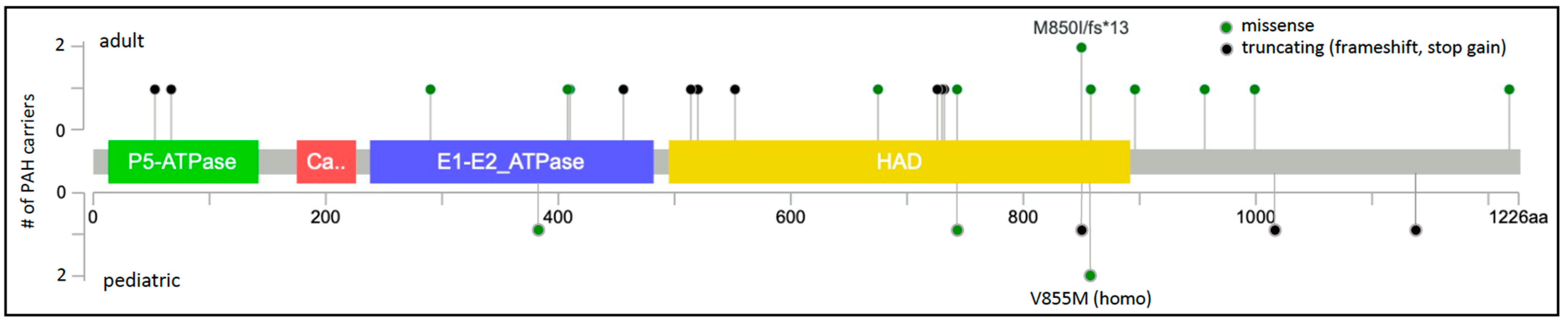
图 2. ATP13A3二维蛋白示意图。
钾双孔结构域通道 ( KCNK3 )
KCNK3基因编码一个双孔结构域钾通道,也称为 TASK1。通过对一个有 5 个受影响成员的家族中的外显子组测序数据进行罕见变异分析,首次将KCNK3鉴定为 PAH 致病基因。随后在至少六项关于 HPAH 和 IPAH 的研究中报告了KCNK3中罕见的错义变异。9种 PAH 相关KCNK3变体的位置如图 3 所示;除了一个变体之外,所有变体都映射到两个保守的离子传输域之一。PAH相关KCNK3的高复发率变异是惊人的,强调了这些残留物在肺血管功能障碍中的重要性。虽然在具有非常早发性严重 PAH 的近亲家族中表明了半显性遗传模式,但所有其他家族的分离数据表明为常染色体显性模式。
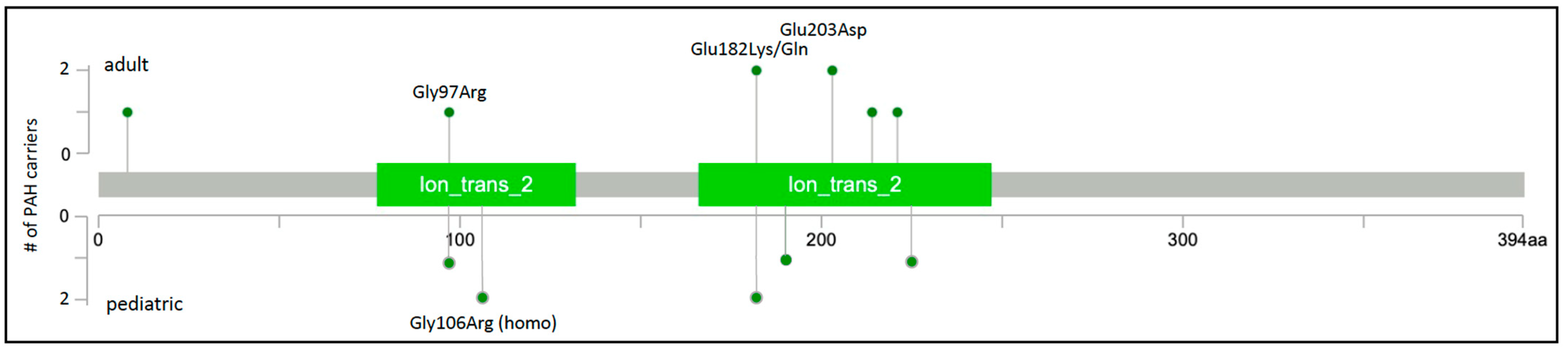
图 3. KCNK3二维蛋白质示意图。
与 PAH 相关的其他通道基因还有其他,例如KCNA5 (OMIM *176267) 编码 Kv1.5,ABCC9 (OMIM *601439) 编码 SUR2,TRPC6 (OMIM *603652),但在 PAH 队列中尚未报道过。
通道基因突变引起PAH及其治疗意义
目前的 PAH 疗法针对血管活性通路来治疗症状和延缓疾病进展,但没有一种是治愈性的。迫切需要旨在抑制或逆转肺血管重塑以阻止疾病进展和提高生存率的新疗法。离子和小代谢物通道在细胞稳态、感知和对局部微环境的变化作出反应方面发挥着重要作用。在肺脉管系统中,由基因变异、药物干预或局部环境因素引起的通道功能变化可以改变血管张力、细胞增殖和细胞代谢。钾离子通道功能障碍已在来自 PAH 患者的 PASMCs 中被记录,其基因变异为ABCC8和KCNK3。改变的 ABCC8在没有已知通道基因变异的 IPAH 病例中也报道了KCNK3表达,这表明这些基因或通道的治疗性调节可能对 PAH 广泛有效。
质膜通道可以用选择性激活剂或通道功能抑制剂进行药物靶向。然而,这样的治疗会导致不希望的副作用。例如,用 SUR1 激活剂二氮嗪治疗的高胰岛素性低血糖患者会出现 PH。此外,PAH 与KCNK3和ABCC8变体的关联是由错义功能丧失变体介导的。通道激活剂可能无法改善严重的功能丧失。因此,未来可能会考虑分子遗传治疗方法,包括在 PASMCs 中添加基因或上调正常等位基因。KCNA5在大鼠模型中的成功基因转移恢复了钾通道电流,使缺氧性肺血管收缩正常化,并改善了 PH。或者,对于具有显性负效应的等位基因,可能需要通过靶向 siRNA 敲除突变的等位基因。这种基因疗法在人类中的应用等待其他疾病的长期安全性和有效性研究。当安全性和耐用性得到解决时,肺和肺血管系统是可接近的组织。
总结及展望
我们对 PAH 遗传结构的理解不断发展,新的候选致病基因、新的遗传变异和其他遗传模式正在被识别。三个通道病基因( ABCC8、ATP13A3和KCNK3)中的遗传变异的作用已在多项独立研究中得到验证,并解释了约 2.7% 的 PAH 病例。ABCC8中的功能缺失错义变体是最常见的(占病例的 1.4%),可能是 H/IPAH 和 APAH 病例的原因。已经证明了ATP13A3的基因剂量效应,双等位基因变异是极早发性重度 PAH 高死亡率的原因。ATP13A3的分子机制PAH 的变异(即功能丧失、功能获得和单倍体不足)尚不清楚。KCNK3中的功能缺失变异(仅限错义)是 PAH 的罕见原因。这些基因在更广泛的 PH 中的作用需要进一步研究。了解通道病的因果机制可能为开发基于通道功能抑制或增强的新疗法提供机会。
参考文献:
Welch CL, Chung WK. Channelopathy Genes in Pulmonary Arterial Hypertension. Biomolecules. 2022 Feb 7;12(2):265. doi: 10.3390/biom12020265. PMID: 35204766.

