Science:一种新的细胞死亡形式—— triaptosis(三磷酸肌醇凋亡)
时间:2024-12-16 06:01:58 热度:37.1℃ 作者:网络
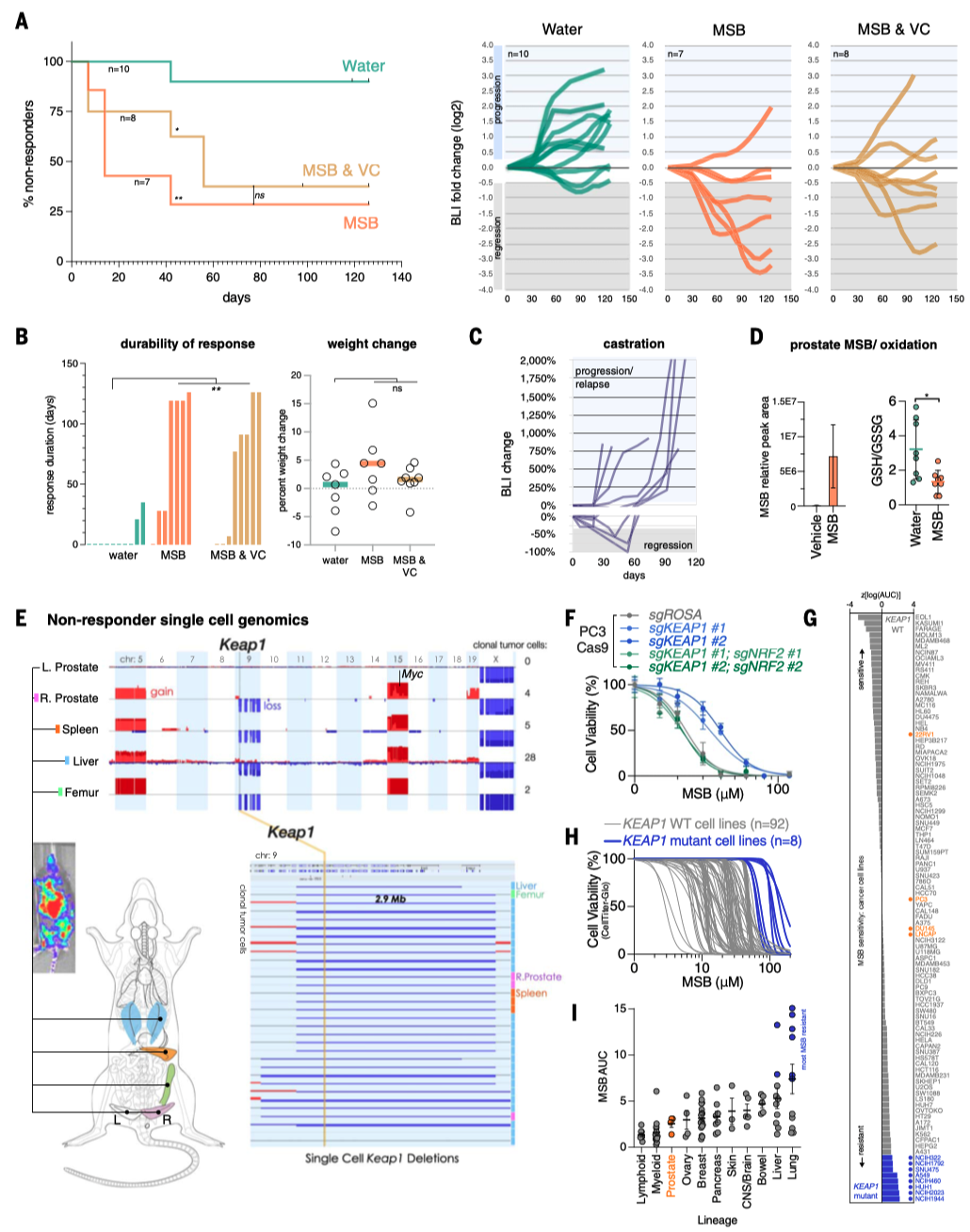
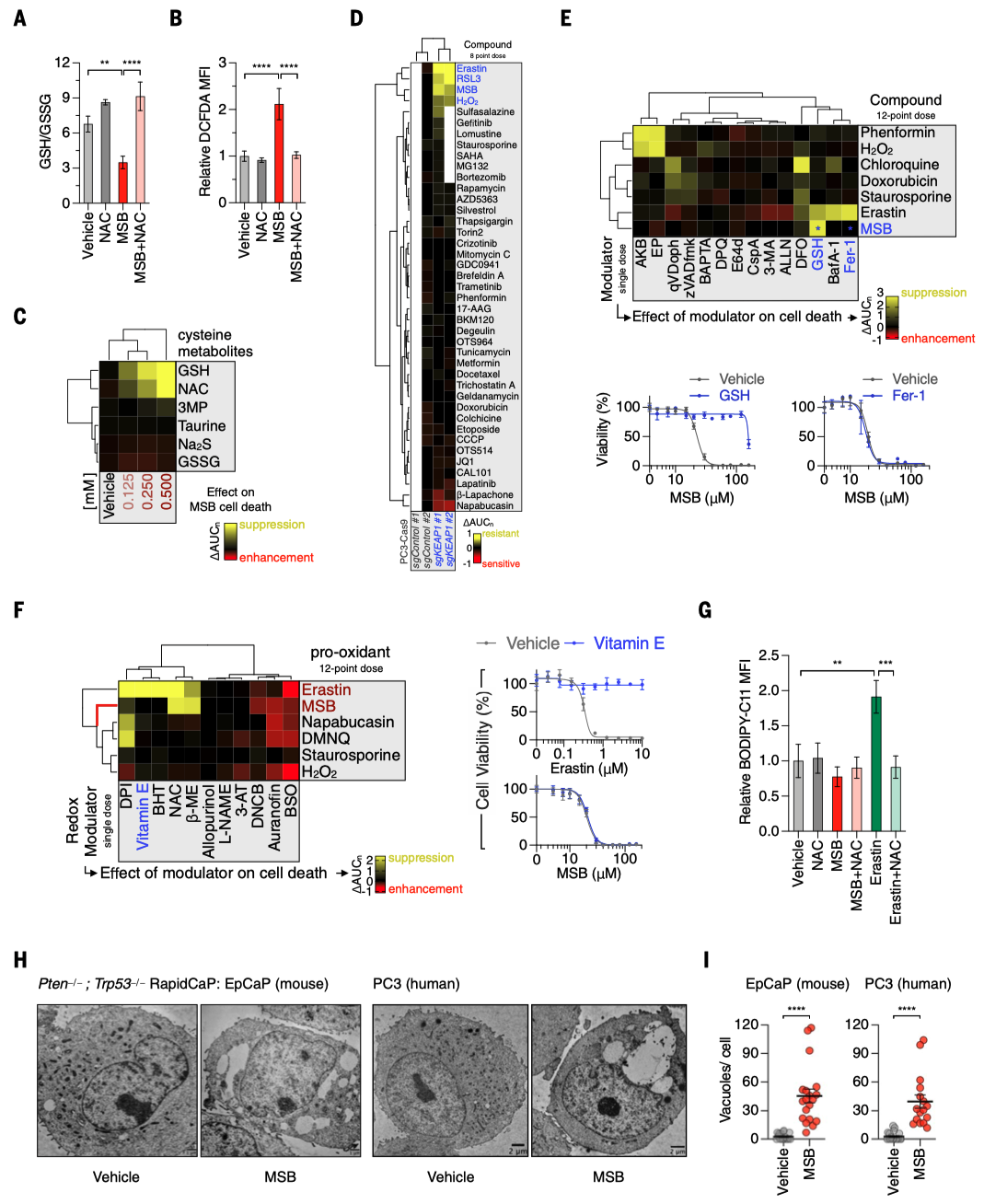
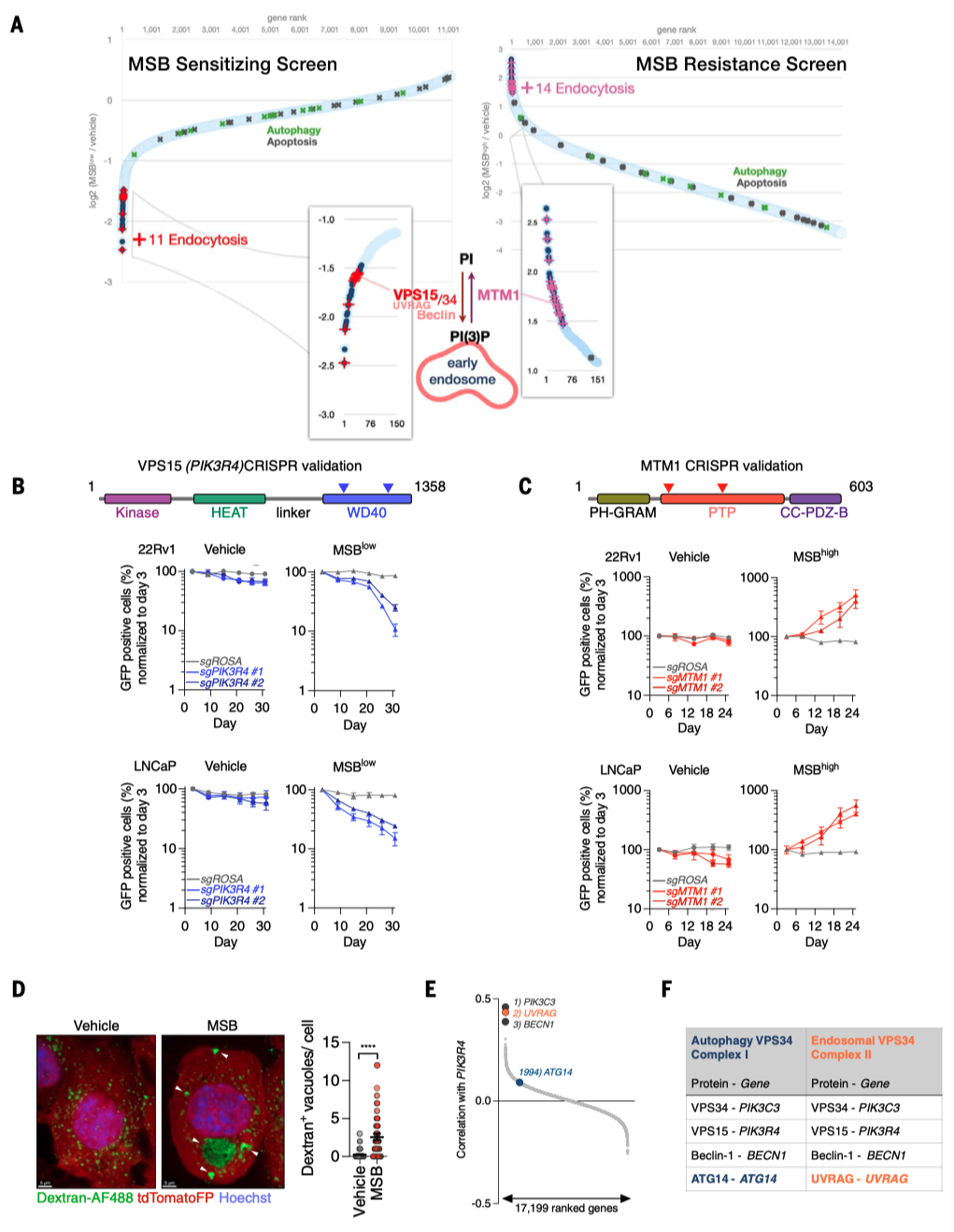
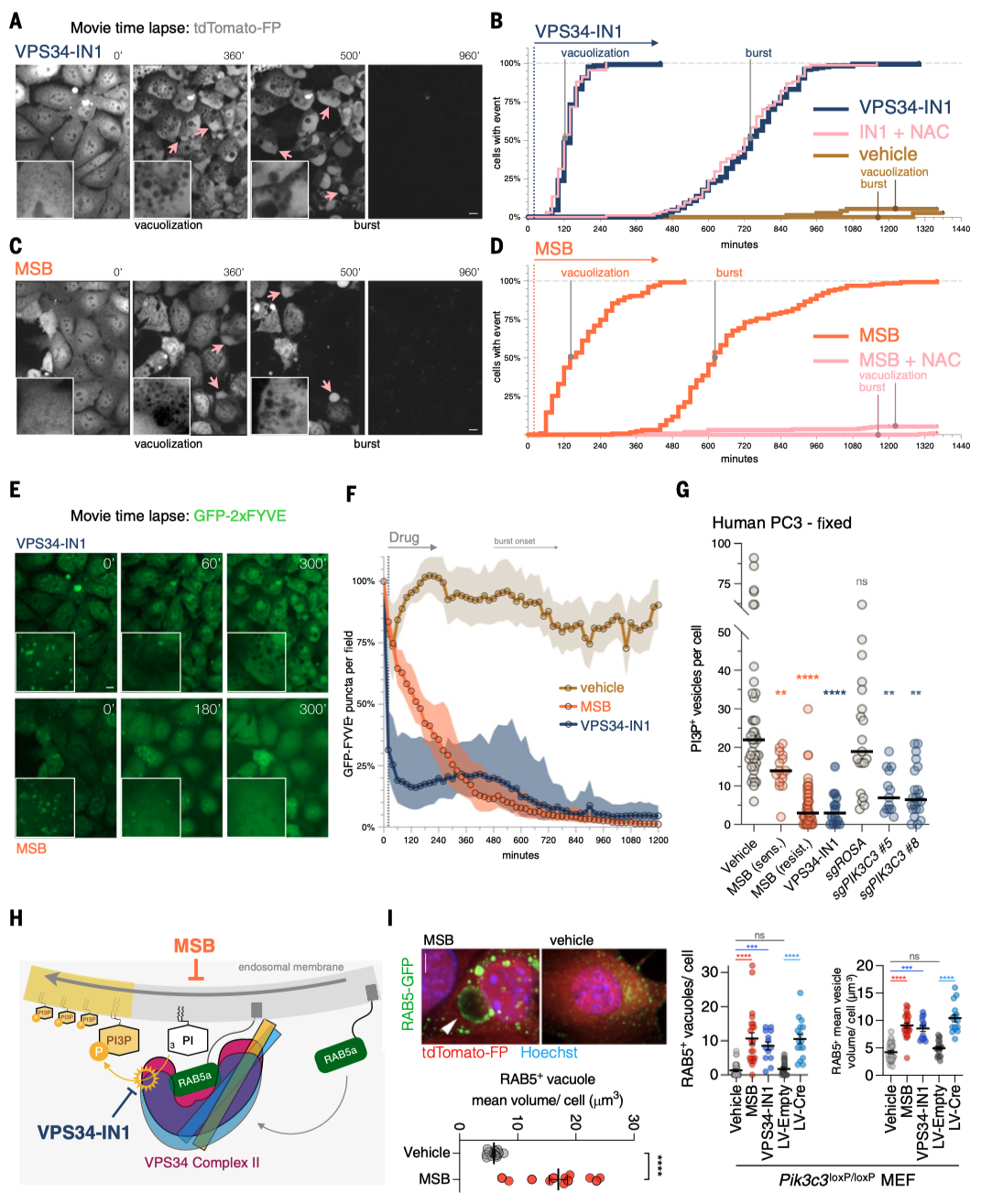
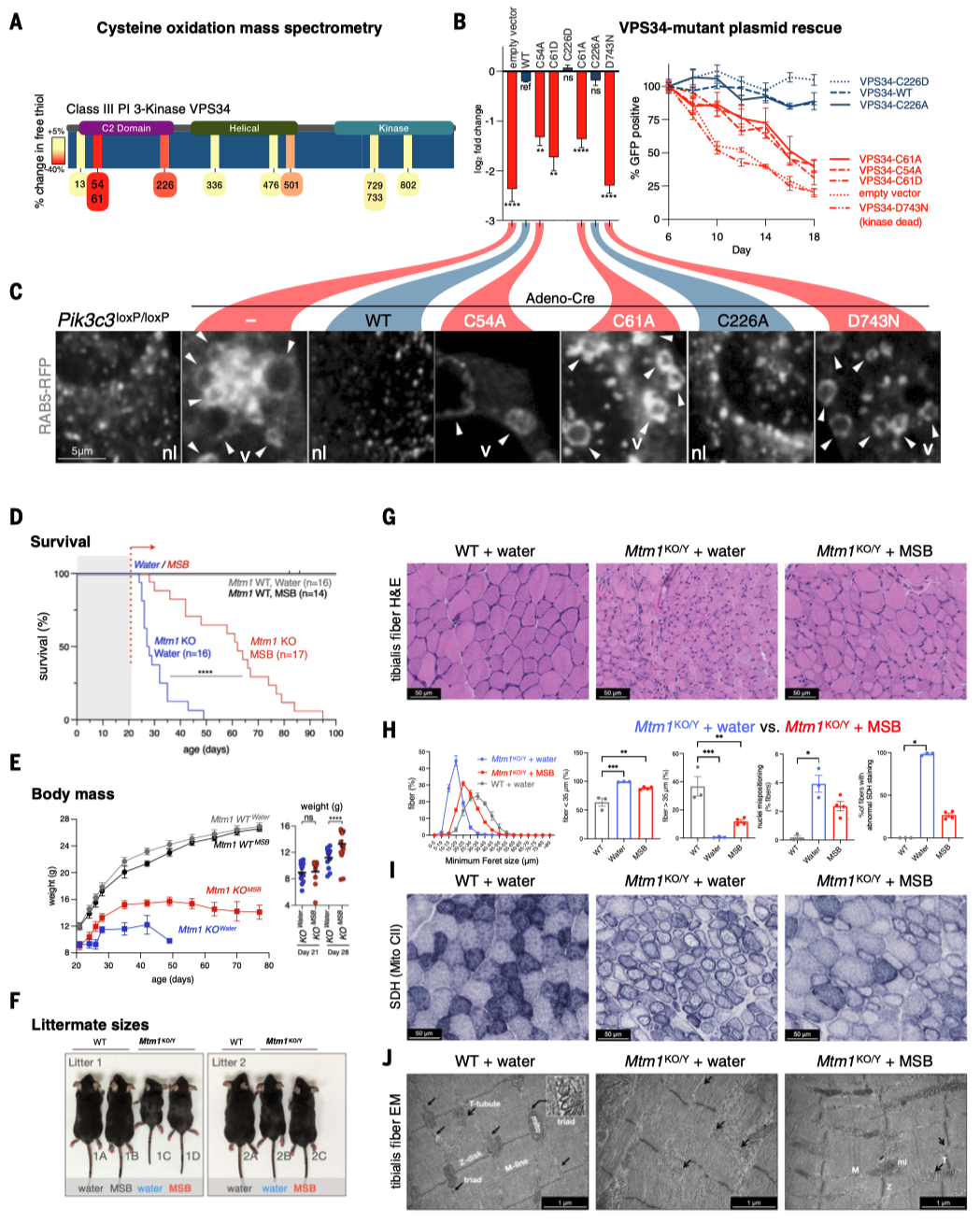
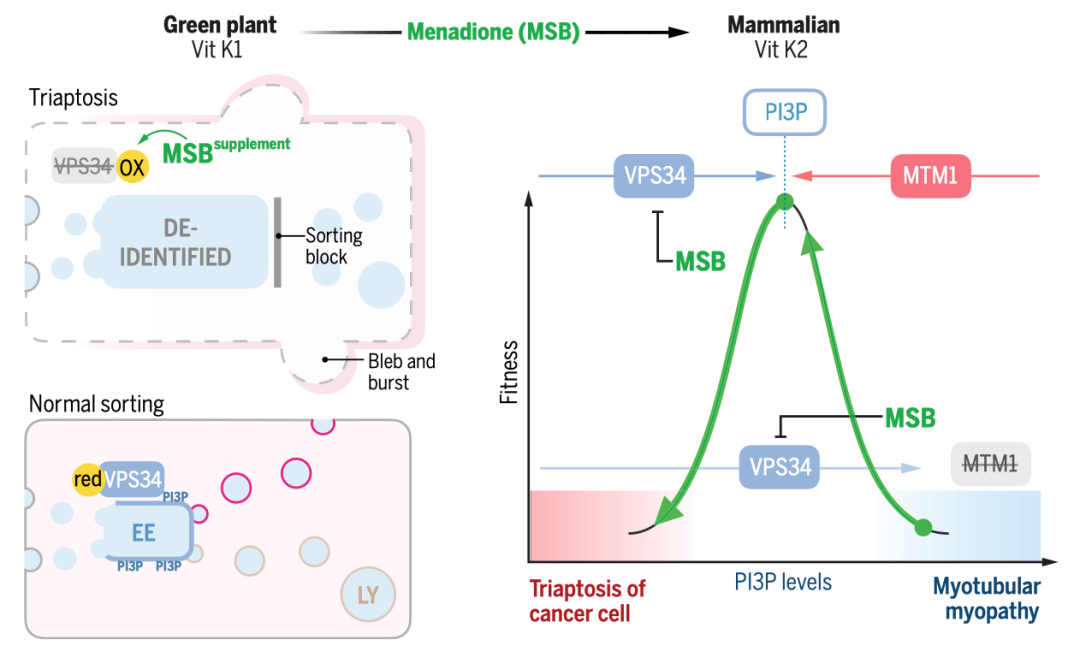
前列腺癌(PC)是男性中最常见的癌症,预计到2024年,美国将有超过299,000例新增病例。这些男性中的大多数将出现缓慢生长的疾病,并可能转变为危及生命的PC,对现有的所有治疗方案产生抗药性。因此,人们非常关注如何通过选择明智的生活方式、饮食和补充剂来减缓疾病进展。这催生了大规模人体试验,包括一项关于膳食抗氧化剂益处的试验:SELECT试验(硒和维生素E预防癌症试验)对35533名健康男性进行了为期10多年的跟踪调查。出乎意料的是,SELECT试验显示,服用抗氧化剂维生素E补充剂的男性患前列腺癌的风险显著增加。抗氧化剂维生素E补充剂对前列腺癌的促进作用立即引出了这样一个问题:抗氧化剂补充剂是否有助于预防前列腺癌?前列腺癌转基因小鼠(GEM)模型为提出这个问题提供了一个平台。 饮食MSB治疗可抑制前列腺癌的发展 首先,我们使用RapidCaP GEM模型来确定前列腺癌是否以及以多快的速度发展为转移性癌症。促氧化剂甲萘醌补充剂(亚硫酸氢钠甲萘醌(MSB))治疗RapidCaP动物,这是一种哺乳动物维生素K的前体,在摄入绿色植物中的植物维生素K后,会在循环中存在。每天在饮用水中补充MSB可以抑制前列腺癌的发展,产生持久的反应。 作者使用了RapidCaP,这是一种将Cre重组酶和荧光素酶转基因体转入PtenloxP/loxP;Trp53loxP/loxP小鼠前列腺的体细胞基因转移方法。这种方法可在完全自然的环境中(4-6周龄)导致局部肿瘤形成,随后发生致死性转移。作者通过每周一次的生物发光成像(BLI)监测前列腺癌的强度和扩散,从而将基因工程小鼠(GEM)模型的严谨性与对全身疾病进展的客观描述相结合。作为促氧化剂,作者测试了两种方法:(1)维生素K前体甲萘醌(也称为维生素K3(VK3)),以水溶性形式给予,即亚硫酸氢钠甲萘醌(MSB);(2)MSB与维生素C(VC)的组合。VC已被证明可在体外和体内氧化癌细胞。在GEM模型中,使用超极化磁共振波谱法证明了PC脱氢抗坏血酸的选择性摄取和细胞内还原作用,并提出了与MSB结合以增强促氧化剂功能。根据BLI判断,25只动物患有原发性和转移性疾病,被随机分为三个试验组:水(10只动物)、MSB(饮用水中浓度为150微克/毫升;7只动物)或MSB加VC(MSB和VC=MSB加1.5毫克/毫升抗坏血酸钠;8只动物)。每天用粉末新鲜配制治疗药物。 对三个组别进行为期18周的每周成像显示,与水对照组病情稳定进展相反,两个治疗组显示出显著的治疗反应、持久的反应以及与水相比无不良体重变化(图1,A和B)。这与RapidCaP的阉割疗法形成鲜明对比,该疗法的治疗反应持续时间少于60天,且随后总是出现阉割抗性,表现为BLI急剧上升和致命疾病进展(图1)。MSB和MSB & VC治疗组中各有一只动物也出现了致死性的1300%和1400%的疾病负担增加,它们被确认为统计异常值。尽管之前有体外协同作用的报道,但在作者的实验中,VC的添加并未改善MSB的治疗效果。作者测量了VC,发现试验动物口服VC后,VC在肝脏和前列腺中的含量并未超过内源性水平。请注意,小鼠与大多数哺乳动物(但不包括人类)一样,能够自行产生VC。这一发现表明,在大鼠体内研究VC的氧化抗癌功效时,高剂量注射VC至关重要,这一点在前列腺癌和结直肠癌的研究中已有报道。相比之下,液相色谱-质谱(LC-MS)分析证实,口服MSB治疗方案可将MSB输送至前列腺,并在那里发挥促氧化剂的作用(图1D)。口服MSB对凝血没有影响。 作者推断,耐药肿瘤可能包含有价值的信息,揭示MSB诱导疾病消退和耐药性的潜在机制。由于RapidCaP系统的原发性肿瘤和转移瘤比人类肿瘤小得多,作者无法直接采集病变进行分子检测。因此,作者转而采用全基因组拷贝数变异(CNA)的单核测序,这是前列腺癌演变的驱动力。通过荧光素酶引导采集疑似肿瘤区域,然后对4′,6-二氨基-2-苯基吲哚(DAPI)染色的细胞核进行荧光激活细胞分选(FACS),作者得以从MSB耐药动物的五个组织中分离出414个单核细胞:左前前列腺(正常对照叶)、右前前列腺(含肿瘤叶)、股骨、脾脏和肝脏。这些样本接受了单核全基因组测序(SNS),以识别和描述癌细胞,正如之前发表的人类前列腺癌样本一样。每个部位的基因组都包含许多肿瘤稀释的假定正常细胞(0% CNA),就像正常的左前前列腺一样。相比之下,右前前列腺(RapidCaP的疾病起源)和荧光素酶阳性的转移部位也含有自发CNA显著(P < 0.0001)增加的细胞(请注意,Cre介导的Pten和Trp53缺失低于作者的CNA检测限600 kb)。这与人类PC中首次显示的进展原理相似。此外,对所有414个单核基因组的分析还揭示了具有重复性CNA的细胞。对60个具有克隆性CNA的癌细胞的聚焦显示,其复杂程度逐渐增加:前列腺<脾脏<肝脏,这可能描绘出转移的路径。如图1所示,在图像浏览器中打开,癌细胞克隆有两个全染色体增益,包括先前发表的15号染色体上的Myc,以及9号染色体上的5个缺失,其中两个是局部的(小于3兆碱基对(Mbp))。其中一个缺失包含Keap1基因,这是一种肿瘤抑制剂和氧化应激的主传感器。 作者证实了Keap1-/-小鼠胚胎成纤维细胞(MEFs)对MSB的耐受性增加,并在人类PC3和LNCaP细胞中使用了CRISPR-Cas9,这与氧化还原缓冲能力的增加一致(图1F)。接下来,作者分别测试了研究所的100个癌细胞系对MSB的敏感性。作者发现,已知存在KEAP1功能缺失突变的细胞对MSB的耐受性最强(图1,G和H在图像查看器中打开),作者对Broad的DepMap 23Q2数据集的基因性表明,MSB耐受细胞系特有的主要遗传依赖性是致癌转录因子NRF2(由NFE2L2编码),该因子受KEAP1抑制。作者使用CRISPR-Cas9证实了这种与MSB的假定联系:人类LNCaP和转移性PC3细胞在KEAP1缺失后对MSB的耐受性增强,但NRF2的共靶向作用会消除这种效应,这与上位遗传关系一致(图1F)。通过对无应答动物的肝脏和肺部转移瘤进行SNS分析,发现了一个携带NRF2(Nfe2l2基因)扩增的转移子克隆。 作者在临床前转移性PC GEM模型中的实验显示:(1)长期口服MSB可显著减缓疾病进展;(2)与RapidCaP中的阉割疗法不同,MSB很少出现致死性耐药性。基因组学显示:(3)耐药性可能由KEAP1-NRF2抗氧化系统控制,这表明癌细胞处于氧化选择压力下。由于人类PC细胞系在MSB敏感性方面排名最高(图1),并且由于KEAP1-NRF2通路在人类PC中很少改变,作者认为MSB应该几乎没有先验遗传抗性。因此,作者试图更好地了解MSB究竟是如何作用于癌细胞的。 MSB触发一种依赖硫醇的细胞死亡模式 对细胞死亡途径的系统分析表明,MSB通过一种独特的氧化细胞死亡机制杀死癌细胞,我们称之为三磷酸肌醇凋亡。我们使用全基因组CRISPR筛选来了解潜在的生物学原理,发现MSB会消耗早期内体(EE)膜脂质磷脂酰肌醇3-磷酸(PI(3)P)。PI(3)P定义了EE区,允许将衍生的囊泡分拣回质膜或进入溶酶体降解系统。 与前列腺中一样(图1D),MSB在体外也导致谷胱甘肽(GSH)耗竭(图2A),并增加细胞质活性氧(ROS)水平(图2B)。因此,作者测试了MSB效应的细胞自主机制。正如所料,细胞存活率与细胞死亡呈负相关,补充谷胱甘肽或其前体N-乙酰半胱氨酸(NAC)可有效防止MSB杀伤,但补充下游半胱氨酸代谢产物则无效。预先培养强细胞渗透性谷胱甘肽乙酯可有效降低细胞对MSB的敏感性,证实还原保护作用发生在细胞内部。为了量化并比较调节剂对MSB杀伤的保护作用和致敏作用,作者计算了标准化曲线下面积的变化(ΔAUCn),并生成按无监督层次聚类排序的ΔAUCn热图。这种方法使作者能够全面、定量地总结MSB-kill的特性。 接下来,作者测试了MSB是否涉及离散的死亡途径,因为之前的文献中提到了多种冗余且相互排斥的细胞死亡途径。根据一些观察结果,作者首先测试了MSB是否通过干扰线粒体电子传递链(ETC)发挥作用:植物K-维生素类叶醌(VK1)在叶绿体电子传递链(ETC)的光系统I中至关重要。泛醌是线粒体电子载体,在结构上与K-维生素类甲萘醌(VK2)有关,Pten和/或Trp53突变PC细胞易受线粒体复合体I抑制的影响。然而,作者发现MSB-kill不受酵母NDI1(一种用于修复线粒体复合体I抑制剂的转基因)的抑制,且MSB不会诱导线粒体ROS。 接下来,作者使用CRISPR衍生的同源KEAP1突变体PC3细胞(图2D),筛选具有已知细胞毒性机制的药物,以确定其中是否有与MSB具有相同特性的药物。对筛选结果进行聚类分析后发现,40种细胞毒性物质中只有3种与MSB相似:H2O2以及两种铁死亡诱导剂erastin和RSL3。此外,作者还证实了MSB和erastin引起的转录组变化之间存在很强的相关性。因此,为了更精确地从药理学角度定义MSB所涉及的细胞死亡途径,作者使用了一种称为调节谱分析(modulatory profiling)的方法。作者触发了三种已知的细胞死亡模式(凋亡、程序性坏死和铁死亡),然后使用各自的途径特异性调节剂阻止了这些模式。将得到的特征与MSB的特征进行了比较。如图2所示,在图像浏览器中打开,泛-半胱氨酸酶抑制剂z-VAD-fmk和qVD-oph可选择性地保护细胞免受staurosporine诱导的细胞凋亡,但不能保护细胞免受MSB-kill。Bax–/–;Bak–/– MEFs 仍然对 MSB 敏感,作者没有发现任何半胱氨酸蛋白酶激活、三磷酸腺苷(ATP)耗竭或自噬体积累。使用替代电子受体(AKB和EP)或聚(ADP-核糖)聚合酶(PARP)抑制剂DPQ以及细胞内钙螯合剂BAPTA-AM补充氧化型烟酰胺腺嘌呤二核苷酸(NAD+)可以抑制H2O2介导的程序性坏死。Erastin诱导的铁死亡可被脂质ROS清除剂ferrostatin-1、铁螯合剂去铁胺(DFO)、溶酶体抑制剂巴法洛霉素A-1或GSH有效抑制(图2)。然而,MSB-kill与上述细胞死亡方式没有任何共同特征(图2)。谷胱甘肽是它与erastin唯一共有的功能性调节剂。这些发现共同表明,MSB通过一种特定类型的氧化应激杀死细胞,而铁死亡的关键调节剂(如ferrostatin-1)对此没有影响。这一点在人类和小鼠来源的PC细胞中得到证实。 作者进一步测试了MSB诱导的氧化应激与使用苯乙福明(线粒体)、氯喹(溶酶体)和阿霉素(细胞核)诱导的氧化应激的分区情况,每种氧化应激都可以通过各自的已知调节剂得到缓解(图2)。然而,这些调节剂都无法抑制MSB的杀伤作用,反之亦然,谷胱甘肽只能阻止MSB的杀伤作用,而不能阻止其他氧化剂引起的死亡。这表明MSB可以针对一种独特的硫醇敏感细胞死亡途径。 为了更好地理解这种硫醇选择性途径,作者测试了11种已知的氧化还原调节剂对MSB毒性和其他四种促氧化剂化合物的影响。如图2F在图像浏览器中打开所示,staurosporine作为11种调节剂的阴性对照化合物。硫代丁二脒(BSO)是一种谷胱甘肽(GSH)生成抑制剂,通常可使细胞对所有五种促氧化剂(erastin、MSB、napabucasin-BBI608、1-chloro-2,4-dinitrobenzene或DMNQ和H2O2)敏感,而DNCB(一种硫氧还蛋白还原酶抑制剂)对MSB的特异性更强。相反,NADPH氧化酶(NOX)抑制剂二苯基碘鎓氯化物(DPI)可抵御五种促氧化剂中的四种,但对MSB的作用较弱。对调节剂进行聚类分析后发现,erastin和MSB之间存在两个关键差异。最值得注意的是,维生素E(α-生育酚)是脂质过氧化反应的特异性抑制剂,它能够阻止erastin引起的铁死亡,但对MSB杀伤没有影响(图2F)。另一种亲脂性抗氧化剂二丁基羟基甲苯(BHT)也能从erastin中选择性解救出来,但不能从MSB中解救出来。这与硫醇还原剂NAC和β-巯基乙醇(β-ME)形成鲜明对比。这些发现已在人源和小鼠来源的PC细胞中得到证实。最值得注意的是,作者发现与erastin相反,MSB不会导致脂质过氧化,这是铁死亡的特征,如使用膜脂质ROS报告基因BODIPY-C11的流式细胞仪分析所示(图2G)。 最后,作者使用透射电子显微镜(TEM)分析了MSB诱导的人类PC3和RapidCaP来源的上皮细胞PC细胞(EpCaP)死亡的形态特征。这揭示了细胞质空泡的大量堆积(P < 0.0001)(图2,H和I);然而,作者没有注意到经典细胞死亡途径的特征,例如染色质凝结。综上所述,作者的分析表明,MSB通过氧化细胞死亡机制杀死细胞,该机制与铁死亡具有相同的生化特征和遗传特征,例如硫醇耗竭和KEAP1依赖性,但与脂质过氧化无关,且形态各异。 全基因组筛选MSB-kill的功能调节因子 为了区分MSB-kill的因果事件和仅相关事件,作者决定(1)使用CRISPR-Cas9筛选,以确定与MSB-kill功能相关的基因;(2)在全基因组范围内进行筛选,以便比通过聚焦途径筛选更客观地揭示潜在原理;(3)同时进行正选择和负选择筛选,从相反的角度解决问题。通过亚致死MSB进行敏化筛选,以确定仅在接触EC10(有效浓度达到10%的效果)时才产生不利影响的基因改变(“MSBlow”;负选择)。还进行了抗性筛选,以确定在高浓度(EC50)下具有生存优势的基因改变(“MSBhigh”;正选择)。两种筛选方法都比较了基因扰动加载体与基因扰动加MSB的适应性效应。作者使用了布罗德研究所的布鲁内洛全基因组单导RNA(sgRNA)文库,该文库包含76441个sgRNA,分别针对19114个人类编码基因(每个基因有四个sgRNA)和1000个对照sgRNA,取自人类转移瘤衍生PC3细胞系。 对敏化筛选(MSBlow)中前沿命中基因的基因本体分析揭示了介导内吞作用的基因(图3A):内吞素A3(SH3GL3),它 介导内吞作用(FEME)的快速内吞素(endophilin-mediated endocytosis),以及巨泡吞噬作用和膜皱褶效应物(ELMO2、NCKAP1、ACTN2和KXD1)。MSB高阳性选择筛选的基因本体分析显示,晚期内体相关基因的富集。这些基因包括RAB7-三磷酸鸟苷(GTPase)效应子,分别与晚期内体运输(WDR91和FYCO1)、成熟(RAB9A、SCARB2和ANXA8)和酸化(CLCN4和ATP13A2)有关,表明这些基因的缺失会增加对MSB的耐受性。这些数据共同表明,MSB对细胞的杀伤与内吞作用存在功能联系。更具体地说,这些数据表明,抑制内吞作用和内体形成的早期步骤可以支持MSB杀伤,而抑制内体形成的晚期步骤则可以抑制MSB杀伤。 作者注意到,VPS15是CRISPR筛选出的主要致敏基因之一(图3A,PIK3R4基因)。这一点之所以突出,是因为VPS15是内体III类磷脂酰肌醇(PI)3激酶VPS34复合物的组成部分。该复合物通过标记细胞内体早期(EE)区室中的磷脂酰肌醇3-磷酸(PI(3)P)脂质来定义该区室。该区室是内吞途径的中央枢纽,因为内吞途径是通过它进行引导的。值得注意的是,MTM1磷酸酶在MSB抗性筛选中处于领先地位。这种磷酸酶是内体VPS34激酶的直接拮抗剂(图3A),因为它可以将PI(3)P还原为PI。这立即表明,MSB-kill的核心可能是一个由酶调节的反应步骤:脂质磷脂酰肌醇3-磷酸(PI(3)P)的产生与消除(图3A)。因此,作者测试了这一假设。 首先,作者使用两种独立的、针对特定基因组的sgRNA,在三种人原代和转移性PC细胞系中验证了这两个筛选结果。竞争分析证实,与对照引导相比,CRISPR靶向VPS15(由PIK3R4编码)使细胞对MSB-kill更敏感。相比之下,MTM1靶向增强了MSB耐受性(图3、B)。接下来,作者使用两种针对激酶域的独立sgRNA,验证了III类PI 3-激酶VPS34(PIK3C3基因)功能在三种PC细胞系中至关重要。MTM1的CRISPR靶向修复了细胞(至少部分)免受PIK3C3缺失的有害影响,这与该激酶-磷酸酶对的酶拮抗作用一致。 VPS34激酶通过复合体I启动自噬,而复合体II则建立内体腔。作者发现MSB抑制了自噬通量,并导致细胞内吞作用停滞,无法摄取细胞外的葡聚糖染料(图3D),这与透射电镜结果(图2)一致。布罗德研究所(DepMap,23Q2)整理的1000多种癌细胞的全基因组CRISPR数据库显示,内体VPS34复合物II的特异性蛋白与VPS15的全基因组相关性最高:PIK3C3,其次是UVRAG和BECN1(图3)。值得注意的是,UVRAG是细胞必需的,并且是内吞复合物(II)所独有的。相比之下,自噬复合物独有的ATG14是非必需的,并且与VPS15无关(图3,E和F)。这与作者的筛选结果一致:尽管涉及硫醇代谢、ROS解毒和氧化还原酶途径的基因改变被证实与MSB协同作用,但自噬、凋亡和其他主要细胞死亡途径的标志基因却并非如此(图3A)。综合这些结果,作者推测内体PI 3-激酶VPS34复合物是MSB-kill的功能靶标(图3A),并且MSB以一种对氧化还原敏感的方式间接或直接控制III类PI 3-激酶VPS34。 内体III类PI 3-激酶VPS34是维生素K3-杀灭的功能靶标 作者首先使用延时显微镜来验证维生素K3通过抑制内体VPS34激酶复合物的氧化作用来杀死细胞的假设。这使作者能够更详细地观察和量化导致细胞死亡的步骤。为此,作者利用RapidCaP(Pten–/–;Trp53–/–)肿瘤(EpCaP细胞)衍生出上皮非迁移性PC细胞,以便在24小时内进行细胞死亡成像。细胞表达tdTomatoFP,代表细胞质,揭示细胞质的明显变化。 首先,作者记录了由III类选择性PI 3-激酶VPS34抑制剂VPS34-IN1引起的细胞死亡。如图4A所示,作者注意到在VPS34-IN1治疗后发生了三个独立事件:细胞质空泡化、持续加剧的质膜(PM)气泡形成以及细胞最终破裂,tdTomatoFP细胞质标记完全消失。为了从统计学上比较这一系列事件,将液泡化的开始和破裂的时间绘制为Kaplan-Meier曲线图。如图(图4B)所示,空白对照组几乎没有细胞破裂和液泡化的现象。值得注意的是,细胞死亡后的液泡化是基因VPS34 Pik3c3缺失后的表型,其他研究和作者在MEFs中使用Cre-loxP系统进行了研究。在激酶抑制剂中添加NAC还原剂不会改变动力学、液泡化的程度或突发事件(图4B)。对MSB的分析(图4,C和D)表明,它与VPS34-IN1诱导的液泡化、气泡化和破裂事件序列非常相似。但与此形成强烈对比的是,NAC还原剂消除了液泡化和PM破裂,使事件序列恢复到背景水平(图4D)。接下来,作者测试了维生素K3是否抑制了III类PI 3-激酶VPS34在细胞内质体上产生PI(3)P标记的功能。图4A至D中成像的细胞还带有GFP-2xFYVE转基因,这是PI(3)P的常用报告基因,使作者能够确定PI(3)P是否受到影响以及何时受到影响。正如预期的那样(图4E),激酶抑制剂VPS34-IN1显著降低了典型的GFP-2xFYVE点状信号,表明PI(3)P阳性囊泡丢失。MSB也有类似但较慢的作用,同样在明显的空泡化之前,如成像和定量分析所示(图4,E和F)。在MSB中添加NAC消除了对GFP-2xFYVE斑点的效应,使其与载体处理无法区分。为了测试细胞死亡表型是否在PTEN和/或TP53突变背景以及PC之外具有普遍性,作者分析了MSB-kill在100种癌细胞系中的结果(图1,H和I),并使用了五种来自不同癌症的敏感细胞类型。这表明,在爆裂之前也会出现液泡化,并且该过程会受到NAC的阻碍。请注意,在固定和渗透时,需要xFP或其他细胞质标记来报告细胞在PM爆裂之前的完整性。PC3细胞中的定量免疫荧光证实,MSB与VPS34-IN1一样,在MSB低和MSB高筛选浓度下均减少了PI(3)P阳性囊泡,并产生弥漫的细胞质GFP-2XFYVE信号(图4G)。CRISPR靶向VPS34激酶与MSB和VPS34-IN1的效应相似(图4G)。 小GTP酶RAB5在早期内体上招募并激活VPS34复合物II,因此作者接下来测试了MSB是否干扰RAB5a的定位。然而,作者发现,MSB诱导的液泡上大量RAB5a的修饰与药物抑制(VPS34-IN1)和分别使用CRISPR-Cas9和Cre-loxP在人类和鼠细胞中靶向PI 3-激酶VPS34(PIK3C3)的遗传学方法(图4I)无法区分。接下来,作者将MSB效应与PIKFYVE抑制进行了比较。PIKFYVE将早期内体上的PI(3)P转化为PI(3,5)P2,以促进内体-溶酶体的进程。PIKFYVE抑制会导致出现以晚期内体蛋白溶酶体相关膜蛋白1(LAMP1)为标志的大液泡。相比之下,MSB或VPS34-IN1处理过的细胞则出现LAMP1阴性的液泡。II类PI 3-激酶在细胞膜上生成PI(3)P。药理学靶向和筛选分析表明,它们对MSB杀伤没有提供主要保护,这与III类激酶VPS34主导的PI(3)P生成相一致。同时,MSB不会干扰PIK3C2A的功能。 因此,作者迄今为止的研究结果支持两个主要结论:MSB可以通过间接或直接拮抗内体PI 3-激酶VPS34来杀死细胞,并且这种调节受氧化还原控制。从机制上讲,MSB诱导的内吞小泡停滞可能是由于两个主要的下游处理途径依赖于内体PI(3)P沉积到RAB5阳性小泡上才能同时检测:回收至质膜需要将囊泡上的PI(3)P转化为PI(4)P,而进入内溶酶体降解途径则需要首先识别PI(3)P,然后将其转化为PI(3,5)P2。因此,作者的数据暗示在识别内体阶段存在氧化还原检查点。 MSB氧化了内体VPS34功能和存活所需的半胱氨酸 为了测试MSB是否氧化PI 3-激酶VPS34,作者从MSB或载体处理过的细胞中免疫沉淀出激酶(值得注意的是,MSB不影响VPS34的水平)。然后,作者对其进行连续的烷基化和还原处理,通过质谱揭示半胱氨酸的修饰状态。如图5A所示,作者恢复了VPS34中总共11个半胱氨酸中的9个,并发现MSB在肽1(Cys54或Cys61)和肽2(Cys226)中导致游离硫醇耗竭,并伴随可逆氧化增加。催化结构域半胱氨酸和阴性对照肽不受影响。肽1(Cys54或Cys61)与C2结构域相关,该结构域控制异四聚体VPS34复合物的形成和功能,并被认为是VPS34翻译后调控的中心。作者使用重组纯化的VPS34复合物在腺苷5′-二磷酸(ADP)Glo激酶检测中证实,全长VPS34、VPS34复合物I(CI)和VPS34复合物II(CII)的活性均被MSB抑制。 然后,作者测试了这些VPS34候选半胱氨酸是否能够控制细胞活力。为此,作者使用VPS34(Pik3c3loxP/loxP)条件敲除的MEF细胞进行了基因敲除-质粒拯救实验。这些细胞稳定地转导了候选VPS34突变体,这些突变体将半胱氨酸替换为丙氨酸或半胱氨酸替换为天冬氨酸。后者是真正的氧化模拟错义突变。感染Cre.GFP慢病毒后,内源性VPS34丢失,VPS34突变体救援质粒的适应性以每种条件下GFP阳性细胞的分数来衡量。图5B(空载体)显示,内源性VPS34重组后,没有替代品,这些细胞会大量减少。相比之下,VPS34野生型(WT)质粒有效地挽救了Pik3c3∆/∆细胞。值得注意的是,所有Cys54和Cys61突变体都无法挽救细胞。这表明(i)这两个半胱氨酸对于VPS34的功能至关重要,并且(ii)它们的氧化可能会使激酶复合物失活。相比之下,Cys226突变体仍然具有功能。 最后,作者通过分析细胞表型评估了这些半胱氨酸的突变是否会导致内体VPS34复合物II失活。如图5C所示,Cys54和Cys61突变体表现出强烈的RAB5阳性液泡形成。因此,这些突变体在没有MSB的情况下产生了MSB杀死的特征。 总之,作者的分析表明,Cys54和Cys61可被MSB可逆地氧化,是内体VPS34功能所必需的。因此,它们是VPS34上进化保守的氧化还原开关的有力候选者,该开关控制由MSB引起的氧化细胞死亡。 膳食维生素K3可延长PI3-激酶VPS34导致的致命疾病患者的寿命 由于MSB与PI 3-激酶VPS34拮抗,作者接下来测试了它是否能够像在PC试验中一样口服给药,从而有效抑制一种涉及该激酶的人类致命疾病综合征。X连锁肌管肌病(XLMTM,OMIM编号310400)是一种男孩遗传性致命疾病,由X染色体上MTM1基因突变引起。由于MTM1磷酸酶是PI 3-激酶VPS34的直接拮抗剂(图3A),这些患者无法抑制PI(3)P的产生。这一点已在患者来源的细胞中得到成功验证。 Mtm1基因敲除(KO)小鼠完全重现了XLMTM最严重的表型,即男婴死亡。如图5D所示,Mtm1缺陷雄性小鼠(Mtm1KO/Y,又称Mtm1-KO)在1个月大时因肌肉生长严重衰竭而死亡,如前所述。相比之下,在饮用水中添加MSB可显著(P < 0.0001)延长总体存活时间,中位数达到62天(图5D)。值得注意的是,MSB的给药方式与癌症试验(图1)相同——在饮用水中,浓度相同。如前所述,幼崽的招募和治疗在断奶后21天开始,此时KO雄性已经表现出疾病症状。作者证实,治疗降低了Mtm1KO/Y小鼠的GSH/GSSG比率。整个试验的体重分析(图5E)显示,与对照组相比,接受MSB治疗的Mtm1KO/Y小鼠体重增长显著(P < 0.0001),尽管在试验第21天时体重没有差异(图5E)。四组接受不同治疗的同窝动物的体型显示,接受MSB治疗的KO动物比未接受MSB治疗的KO同窝动物体型更大(图5F)。 对未经处理的Mtm1KO/Y胫骨前肌(TA)的组织学分析显示,与野生型肌肉相比,先前报道的肌纤维萎缩和细胞器定位错误(图5G)以及纤维峰值直径变小(图5H)的情况仍然存在。与未经MSB处理的KO动物相比,Mtm1KO/Y小鼠的TA肌纤维尺寸在MSB处理后有所增加,表现为纤维数量增加,直径变小(<35 μm),而直径较大的纤维数量减少(>35 μm;图5H)。在4%的Mtm1KO/Y纤维中观察到核错位的典型特征,在接受MSB治疗的KO小鼠中,该比例下降至2.4%(图5)。异常细胞外线粒体(通过琥珀酸脱氢酶(SDH)和NADH染色显示;图5,H和I)在94%未经治疗的Mtm1KO/Y小鼠中可见,而在MSB治疗的Mtm1KO/Y小鼠中减少到27%。因此,MSB治疗Mtm1KO/Y小鼠改善了TA肌纤维的几种组织病理学缺陷。通过透射电子显微镜(TEM)对肌肉超微结构进行分析,证实了Mtm1 KO的表型(图5J):肌节杂乱无章,Z盘错位,M线无法检测,肌纤维之间没有线粒体。虽然未治疗的Mtm1KO/Y小鼠体内存在T管,但三联体不存在。在MSB处理的Mtm1KO/Y小鼠中,肌节排列得到改善,线粒体定位于肌纤维之间,与WT肌肉类似。T管也可见,但三联体没有恢复。总体而言,在Mtm1KO/Y小鼠中,肌肉组织学和肌纤维超微结构在MSB给药后得到改善。 开放式笼子监测显示,MSB改善了活动时间、行进距离和平均速度,但Mtm1KO/Y小鼠的后肢数量仅在MSB治疗时与WT没有显著差异。正如预期的那样,在探索模式上没有显著差异。为了更详细地研究临床参数的发病情况,作者进行了第二次试验,发现MSB治疗显著延迟了后肢瘫痪的发病时间(P < 0.001)。最后,作者使用临床评分法评估疾病进展,发现与水相比,接受MSB治疗的Mtm1KO/Y组有显著(P < 0.01)改善(。综合来看,作者的数据表明,MSB介导的PI 3-激酶VPS34靶向治疗可用于有效抑制小鼠这种致残性遗传疾病。 研究结果表明,膳食中添加维生素K3可以成为多种疾病新疗法的基础。这是因为维生素K3是PI 3-激酶VPS34的氧化拮抗剂,而PI 3-激酶VPS34是产生磷脂PI(3)P的酶。在PC细胞中,氧化应激会降低PI(3)P,导致细胞因三磷酸肌醇凋亡而死亡。根据我们的数据,我们推断正常细胞具有足够的还原能力来抵御这种伤害。在肌管肌病中,甲萘醌可以抑制不受抑制的VPS34激酶活性,使PI3P恢复到可以改善肌肉发育的水平。总的来说,我们的发现有助于人们逐渐了解促氧化剂的选择性,并表明确定它们作用的途径可以带来意想不到的治疗机会。 原始出处: Swamynathan MM, Kuang S, Watrud KE, Doherty MR, Gineste C, Mathew G, Gong GQ, Cox H, Cheng E, Reiss D, Kendall J, Ghosh D, Reczek CR, Zhao X, Herzka T, Špokaitė S, Dessus AN, Kim ST, Klingbeil O, Liu J, Nowak DG, Alsudani H, Wee TL, Park Y, Minicozzi F, Rivera K, Almeida AS, Chang K, Chakrabarty RP, Wilkinson JE, Gimotty PA, Diermeier SD, Egeblad M, Vakoc CR, Locasale JW, Chandel NS, Janowitz T, Hicks JB, Wigler M, Pappin DJ, Williams RL, Cifani P, Tuveson DA, Laporte J, Trotman LC.Dietary pro-oxidant therapy by a vitamin K precursor targets PI 3-kinase VPS34 function,Science . 2024 Oct 25;386(6720):eadk9167. doi: 10.1126/science.adk9167