Nature Methods:核苷酸转换模型(Nucleotide Transformer, NT):解锁基因组预测的新纪元
时间:2024-12-02 15:02:03 热度:37.1℃ 作者:网络
引言
在生命科学的领域,DNA曾被誉为生命的“天书”,承载了生物体所有的遗传信息。然而,如何解读这本“天书”,从中提取关键的生物学信息,一直是科学界面临的巨大挑战。从基因表达到增强子活性,再到与疾病相关的遗传变异,这些复杂的分子表型背后隐藏着大量的规律和秘密。但受限于标注数据的稀缺性以及传统方法的局限,基因组学在精准预测和任务迁移性上始终存在瓶颈。
近几年,人工智能(AI)领域的基础模型为这一难题带来了新曙光。这些模型(如BERT、GPT)通过从文本中学习语言规则,在自然语言处理中实现了从无到有的认知飞跃。受此启发,研究人员将这一技术嫁接到基因组学中,开发出了专门用于DNA序列分析的核苷酸转换模型(Nucleotide Transformer, NT)。它不仅能像阅读文本一样解读DNA序列,还能够预测基因组中的关键功能区域。(11月28日 Nature Methods “Nucleotide Transformer: building and evaluating robust foundation models for human genomics”)
更令人兴奋的是,NT模型通过跨物种的数据训练,展现了强大的任务适应性和功能泛化能力。无论是剪接位点的精确定位,还是复杂增强子活性的预测,这一模型都打破了传统方法的桎梏,为基因组学研究提供了前所未有的洞见。

NT模型的诞生:深度学习与大数据的结合
数据是基石:为模型训练提供多样化的资源
NT模型的成功离不开其背后的庞大数据基础。研究团队从以下三种数据集中提取序列信息,为模型提供了多样化和高覆盖度的训练资源:
1. 人类参考基因组:作为模型的起点,提供了统一的标准序列,帮助模型捕捉人类基因组的基础特性。
2. 1000基因组计划数据:整合了3202个人类基因组,涵盖了全球范围内的遗传多样性,从而增强了模型对个体差异的识别能力。
3. 850个物种基因组:跨越从简单模式生物(如酵母)到复杂哺乳动物的数据集,其中包含了11种经典模型生物,为模型的跨物种学习奠定了基础。
这些数据集不仅大幅提升了模型的泛化能力,还让它能够在跨物种任务中表现出色。值得一提的是,数据的多样性对模型的成功起到了关键作用。例如,多物种数据集使得NT模型可以捕捉到保守的基因组特征,而这些特征在功能预测中尤为重要。
自监督学习:解锁基因组中的隐藏规律
NT模型采用了掩码语言建模(Masked Language Modeling, MLM)任务作为训练方法。与NLP领域中的BERT模型类似,NT模型通过随机遮盖DNA序列中的某些核苷酸,并要求模型根据上下文推测被遮盖部分。这种训练方式的优势在于,它不需要大量人工标注数据,却能从未标注的DNA序列中提取深层次的模式。
通过这种自监督学习方法,NT模型成功地在数万亿个核苷酸中发现了DNA的“语言规则”,并提炼出通用的特征表示。这为后续的多任务预测奠定了坚实基础。
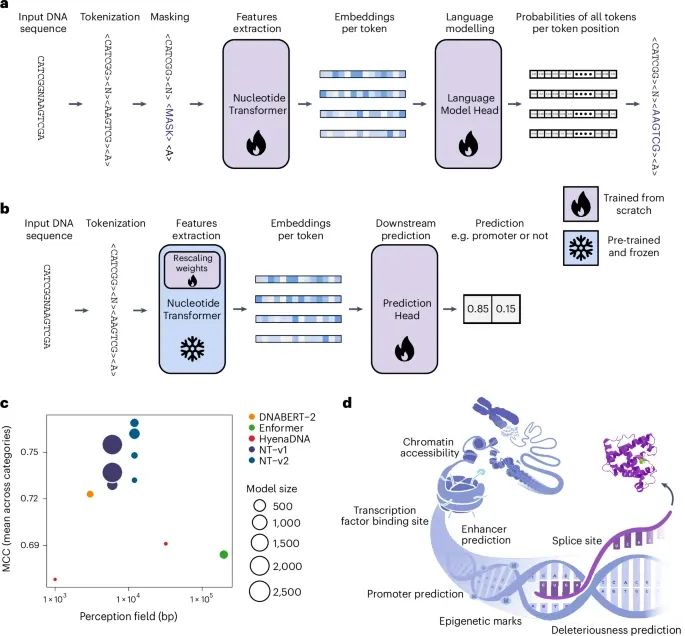
核苷酸转换模型(Nucleotide Transformer, NT)的核心方法以及与其他基因组学基础模型的比较(Credit: Nature Methods)
NT模型的训练和下游任务应用流程 (a) NT训练的概览:展示了NT模型从预训练到微调的完整流程。在预训练阶段,模型通过掩码语言建模(Masked Language Modeling, MLM)任务从未标注的DNA序列中学习核苷酸的上下文依赖特性。(b) 微调(Fine-tuning)的应用:展示了如何通过微调将NT模型应用于基因组预测的下游任务,例如剪接位点预测、增强子活性预测等。在微调过程中,模型的权重被重新调整以适应具体的任务需求。探针测试(Probing):对比了探针测试与微调的区别。探针测试类似于微调,但不对模型权重进行缩放调整(rescaling),以评估预训练模型的通用性。
NT模型与其他基因组学基础模型的比较 (c) 感知野大小(Perception Field Size)、参数数量和性能的对比:图表中比较了NT模型与其他基因组学基础模型(如DNABERT、HyenaDNA和Enformer)的关键指标:
感知野大小:NT模型的感知野更大,能够捕捉更长距离的序列依赖性,这在基因组学中尤为重要。
参数数量:NT模型的参数数量多样化(从500M到2.5B),使得它能够平衡性能与计算资源需求。
性能表现:在18个精心挑选的下游任务中,NT模型在大多数任务中优于其他模型,表现出卓越的跨任务预测能力。
下游任务中考虑的基因组特征 (d) 基因组特征的图示,直观展示了用于下游任务的主要基因组功能区域和特征,包括外显子、内含子、剪接位点、增强子、启动子、组蛋白修饰等。这些特征是基因组功能预测的核心元素。图示的部分来源于其他研究,经过适当改编以适配NT模型的任务需求。
NT模型的多任务能力:解码DNA的多样化功能
研究团队将NT模型应用于18个核心基因组学任务,涵盖剪接位点预测、启动子识别、增强子活性分析和组蛋白修饰等多个领域。通过系统的对比实验,NT模型在大多数任务中都超越了传统模型。
剪接位点预测:让RNA的“拼接”更高效
RNA剪接是基因表达的重要步骤,剪接位点的准确识别对理解基因调控机制至关重要。NT模型在GENCODE数据集上的表现令人赞叹:多物种2.5B模型在6 kb的输入序列中实现了95%的Top-k准确率,并在精确召回曲线(Precision-Recall AUC)上达到了0.98,这一表现远超传统模型SpliceAI-6k。
这一结果表明,NT模型不仅能够高效识别剪接受体和供体位点,还能够在数据稀缺的情况下维持卓越的预测能力。这一特性特别适合应用于需要高精度的疾病研究。
启动子预测:基因“开关”的精准定位
启动子是基因表达的关键调控区域。NT模型在预测启动子时,展现出了极高的准确性:TATA盒启动子预测的MCC为0.76,显著优于传统卷积网络BPNet(MCC为0.68)。
NT模型的成功表明,它不仅能识别启动子的存在,还能精确区分不同类型的启动子,为基因调控研究提供了更强的工具。
增强子活性预测:基因表达的幕后推手
增强子是调控基因表达的重要“开关”。在果蝇S2细胞增强子活性任务中,NT模型表现尤为突出:发育性增强子预测的AUC达到0.75,超过了DeepSTARR模型的AUC(0.74)。转录因子结合位点的突变效应预测:在Dref位点的突变预测中,NT模型的准确率高于其他模型,提升约5%。
组蛋白修饰预测:揭示DNA的包装奥秘
在染色质特征预测任务中,NT模型通过识别组蛋白修饰区域,为表观遗传学研究提供了新的工具:多物种模型的平均AUC为0.95,与DeepSEA模型(AUC为0.96)接近,但NT模型显著降低了计算成本。
这些结果表明,NT模型不仅能够高效预测基因组功能,还能在多种不同任务中实现跨领域的出色表现。
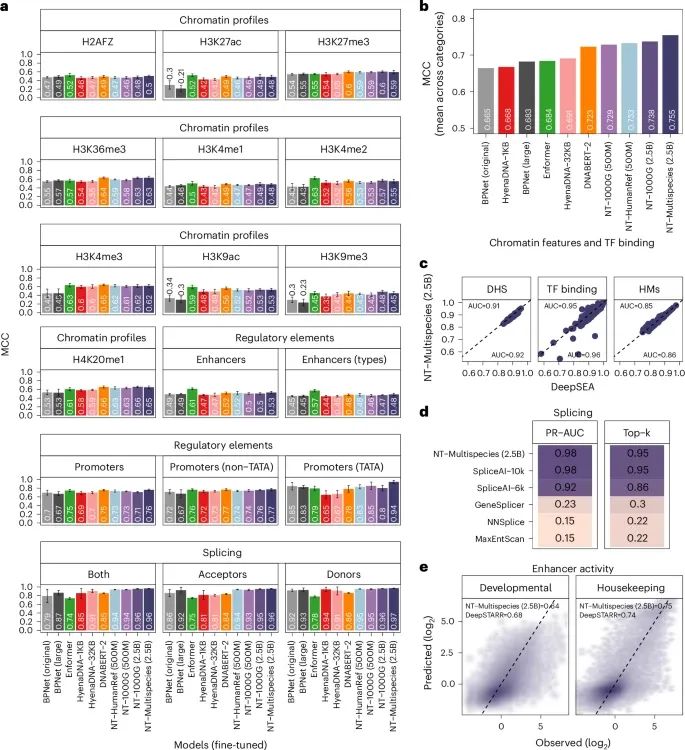
核苷酸转换模型(Nucleotide Transformer, NT)与其他基因组学基础模型的比较(Credit: Nature Methods)
下游任务的整体性能比较 (a) NT模型与其他模型的MCC表现:图中对比了NT模型、HyenaDNA、DNABERT、Enformer以及从头训练的BPNet模型在多个基因组学下游任务中的表现。使用MCC(Matthews相关系数)作为主要评价指标,数据通过十折交叉验证获得(n=10),以均值±2倍标准差(mean MCC ± 2 × s.d.)的形式呈现。NT模型(特别是多物种2.5B参数版本)的表现显著优于其他模型,特别是在感知野较大的任务上,例如剪接位点预测和增强子活性预测。
按任务类别划分的标准化MCC性能 (b) 标准化MCC的对比:不同模型在多个下游任务类别中的平均性能进行了归一化比较。结果显示,NT模型在所有任务类别中都具有卓越的性能,尤其在复杂的序列功能预测(如增强子和组蛋白修饰)中,表现出了明显的优势。
在具体任务中的性能表现
(c) NT模型在DHS、HMs和TF位点预测中的表现:针对人类细胞和组织的DNase I高敏感位点(DHSs)、组蛋白修饰标记(HMs)以及转录因子结合位点(TF sites)的预测,NT模型的ROC曲线下面积(AUC)与基线模型DeepSEA相比显著提高。每个点代表一种基因组特征的AUC,NT模型的平均AUC值明显高于DeepSEA。
(d) 剪接位点预测的对比:在人类基因组的剪接位点预测中,NT模型的性能与SpliceAI以及其他剪接模型进行了比较。多物种2.5B NT模型的表现与SpliceAI相当,且在某些任务上有所超越,尤其是在6 kb感知野范围内的预测精度上。
(e) 增强子活性预测的表现:NT模型在果蝇S2细胞中预测发育性和管家型增强子活性时,与DeepSTARR模型进行了对比。多物种2.5B NT模型在预测AUC上显著超越DeepSTARR,尤其在发育性增强子活性任务中展现了卓越的性能。
模型解析:NT如何“读懂”DNA的语言?
层级注意力机制:精准聚焦基因组关键区域
NT模型的Transformer架构通过层级注意力机制(Hierarchical Attention Mechanism),能够聚焦于基因组的关键区域。例如:外显子和内含子区域,模型注意力的集中度高达117个,展现出对功能区域的强大识别能力。增强子区域,NT模型几乎完全覆盖了该区域,突显了其在功能区域捕捉中的高效性。
这些能力让NT模型在不同任务中能够灵活调整注意力焦点,从而精准完成多样化的预测任务。
零样本学习:从未见过的数据中预测功能
NT模型的另一个亮点是其零样本学习能力(Zero-shot Learning)。即使没有明确的标注,模型也能推测DNA变异的潜在功能,在预测终止变异的功能重要性时,NT模型对终止变异的分数显著低于其他类型的变异,表明它能够准确评估变异的致病风险。
这种能力让NT模型在疾病相关变异的筛选中表现出强大的潜力,尤其是在缺乏标注数据的情况下。
NT模型的优化与未来应用
NT-v2:更小、更快、更强
为了进一步提高效率,研究团队开发了NT模型的优化版本NT-v2。这一版本在保持高性能的同时,显著减少了计算资源需求:参数数量减少至1/50,但性能依然卓越,250M参数模型的平均MCC达到0.769。上下文长度扩展至12 kb,能够捕捉更长距离的基因组依赖关系。
这些优化让NT-v2模型特别适合资源受限的应用场景,为基因组学研究带来了更多可能性。
应用前景:赋能精准医学和多组学研究
NT模型的应用潜力十分广泛,包括:
1. 疾病预测和致病变异筛选:NT模型能够帮助快速识别潜在的致病变异,为精准医疗提供基础。
2. 跨物种基因功能研究:NT模型对跨物种的适应性使得它能够揭示更多基因功能的本质。
3. 多组学数据整合:NT模型能够整合基因组学、转录组学和表观遗传学数据,探索复杂的生物网络。
随着技术的进一步发展,NT模型有望在医学研究和基础科学领域发挥越来越重要的作用。
核苷酸转换模型(Nucleotide Transformer, NT)以其大规模数据处理能力和精准的预测性能,为基因组学研究注入了全新的活力。从解码DNA语言到预测基因功能,它在多任务中的表现令人信服。未来,随着NT模型的不断优化和应用探索,它将在精准医学、多组学整合和基础生命科学研究中扮演更加重要的角色,开启基因组学研究的智能化新时代。
参考文献
Dalla-Torre, H., Gonzalez, L., Mendoza-Revilla, J. et al. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nat Methods (2024). https://doi.org/10.1038/s41592-024-02523-z

