东京大学Hiroaki S.课题组NC论文:体外筛选大环肽的SARS-CoV-2主要蛋白酶抑制剂
时间:2023-08-16 11:46:19 热度:37.1℃ 作者:网络
天然产物中存在的γ-氨基酸在其生物活性中发挥着重要作用。γ-氨基酸不仅能诱导肽链形成独特的二级构象,还能提高肽链水解的稳定性和细胞渗透性,含有γ-氨基酸的大环肽是一类结构新颖且具有吸引力的治疗性多肽。大环肽作为一类新兴的治疗药物,可以调节蛋白质之间的相互作用。与传统小分子药物的自动化高通量筛选系统相比,大环肽可以通过核糖体翻译合成,并可以使用体外筛选技术进行识别,从而提高筛选效率。
然而,核糖体将γ-氨基酸插入到多肽中十分具有挑战性。γ-氨基酸作为核糖体延伸单元的难点在于其自身容易发生环化反应,东京大学Hiroaki Suga课题组使用构象限制的环状γ2, 4氨基酸(cγAAs) 来避免自去酰化的问题,并利用其课题组前期开发的随机非标准肽集成发现 (The Random nonstandard Peptides Integrated Discovery,RaPID) 系统从而高效筛选与SARS-CoV-2主要蛋白酶具有高亲合力的含γ-氨基酸的大环肽。相关工作以“In vitro selection of macrocyclic peptide inhibitors containing cyclic γ2, 4-amino acids targeting the SARS-CoV-2 main protease”为题发表在Nature chemistry期刊上 (Nat. Chem., 2023, 15: 998-1005)【1】。
核糖体合成包含非天然氨基酸 (N-甲基-L-α-氨基酸,D-α-氨基酸和β-氨基酸) 多肽的技术在建立合成多肽库以及发现新药方面起到了强有力的推动作用。但是γ-氨基酸的延伸仍然是一项艰巨的挑战,γ-氨基酸掺入困难主要有两个原因,其一是γ-氨酰-tRNA的自去酰化反应。其二是由于催化环境的兼容性较差,tRNA上A位点的γ-氨基和肽基-tRNA P位点上的酰基之间成键速度缓慢。为了解决上述问题,作者利用构象限制的环状γ2, 4氨基酸避免自去酰化问题 (图1a),同时作者通过优化t-RNA的T-stem和D-arm的序列得到工程t-RNAPro1E2,从而促进t-RNA与延伸因子EF-Tu和EF-P的结合,以加速肽基的转移。并且作者利用前期开发的Flexizyme催化cγAAs和t-RNAPro1E2形成氨酰t-RNA (cγAA-tRNAPro1E2),以及利用可调节EF-Tu和EF-P浓度的灵活的体外翻译 (the Flexible In vitro Translation, FIT) 系统将非天然氨基酸引入至肽链中从而完成含cγAAs肽链的合成。
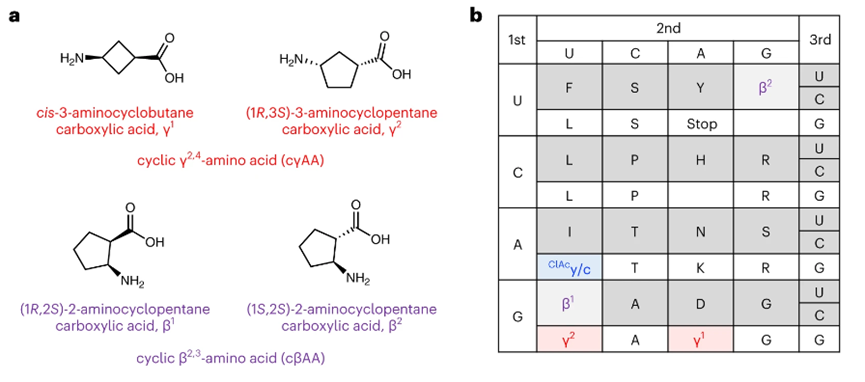
图1: (a) γ2, 4氨基酸以及环状β2, 3氨基酸的结构;(b)γ1, γ2, β1, β2对应的重编密码子表。
为了构建含有环状γ2, 4氨基酸以及环状β2, 3氨基酸的大环肽库,作者首先利用flexizymes将不同的氨基酸预先加载到各自的t-RNA上,然后通过FIT系统分别利用不同的t-RNAPro1E2将γ1, γ2,β1, β2分配到GAG, GUG, GUU, UGU密码子上 (图1b)。同时为了通过硫醚键使肽链环化,分别在启动子AUG密码子和延长子AUG密码子处通过不同的t-RNAPro1E2引入N-氯乙酰-D-酪氨酸 (ClAcy)和D-半胱氨酸 (c)。随后,作者利用该课题组前期开发的随机非标准肽集成发现 (RaPID) 系统构建含有cγAAs和cβAAs的大环肽库。在快速筛选系统的mRNA文库制备中,在每个mRNA的3’末端与嘌呤霉素 (Pyromycin) 连接子结合,嘌呤霉素部分与翻译肽的C-末端结合 (肽的大环化发生在N端ClAc部分和Cys的硫醇之间),于是即可产生与mRNA“基因型”相对应共价连接的表达肽“表型”,并将mRNA反转录成cDNA,得到肽/mRNA/cDNA偶联物 (图2)。随后,作者挑选SARS-CoV-2主要蛋白酶作为测试靶蛋白,以期开发有效的非共价结合于活性位点的Mpro抑制剂。
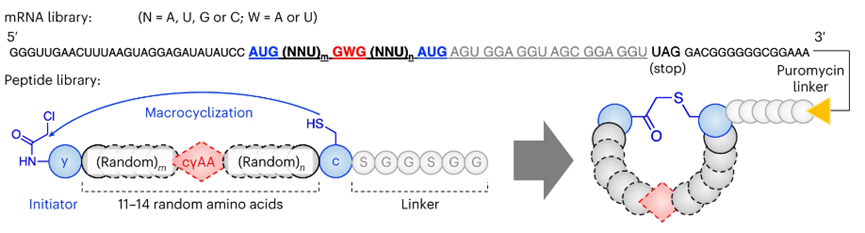
图2: 随机mRNA文库及相应肽库的构建
作者,首先用裸磁珠处理肽库,去除与磁珠非特异性结合的偶联物,然后针对固定在磁珠上的Mpro蛋白对肽mRNA融合文库进行筛选。将富集得到的环肽的mRNA通过逆转录和PCR扩增,然后转录到下一代mRNA文库中。重复上述操作直至回收率不再增加,编码肽的mRNAs被富集,其序列可以通过相应cDNA测序来确定。通过对cDNA文库的测序发现文库中含有多个含有cγAAs的多肽家族,并从中选取并合成7个含有cγAAs的大环肽 (GM1-GM7) 进一步分析其结合亲合力、抑制活性和蛋白水解稳定性。其中GM1、GM2、GM4和GM6序列中含有γ1残基,GM3、GM5和GM7含有γ2残基。
作者,首先通过表面等离子体共振 (SPR) 评估GM1-GM7与Mpro的亲合力。其中,N端具有保守残基yFHγX的环肽GM1、GM4和GM5表现出很强的亲合力,KD值分别为2.3、5.2、5.2 nM。为了评估cγAA残基对效价的贡献,作者用Ala取代cγAA后,发现GM1γ14A、GM4γ14A和GM5γ14A的亲和力下降了一个数量级,表明保守的yFHγX基序中cγAA残基对结合的重要性。随后,作者评估了GM1-GM7对Mpro的水解活性的抑制剂活性,发现含有保守基序的GM1、GM4和GM5表现出较强的抑制活性,其IC50分别为40、50和50 nM,表明IC50和KD值之间存在相关性。同样,作者也测定丙氨酸突变体的抑制活性。值得注意的是,GM1γ14A的抑制活性完全丧失,但GM4γ14A和GM5γ14A却保留了抑制活性,作者认为可能存在其他调节因素 (图 3a)。接下来,作者评价了含cγAA环肽及其突变体在人血清中的半衰期,发现含有cγAA残基的环肽相比于Ala突变体表现出较高的肽酶抗性,表明存在立体位阻的cγAA残基表现出更高的血清稳定性 (图 3b)。
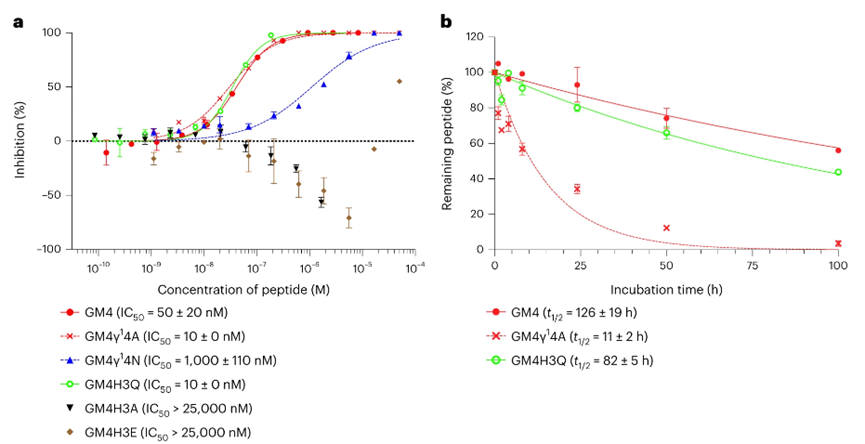
图3: (a) 大环肽抑制Mpro的剂量效应分析;(b) 大环肽血清稳定性测定
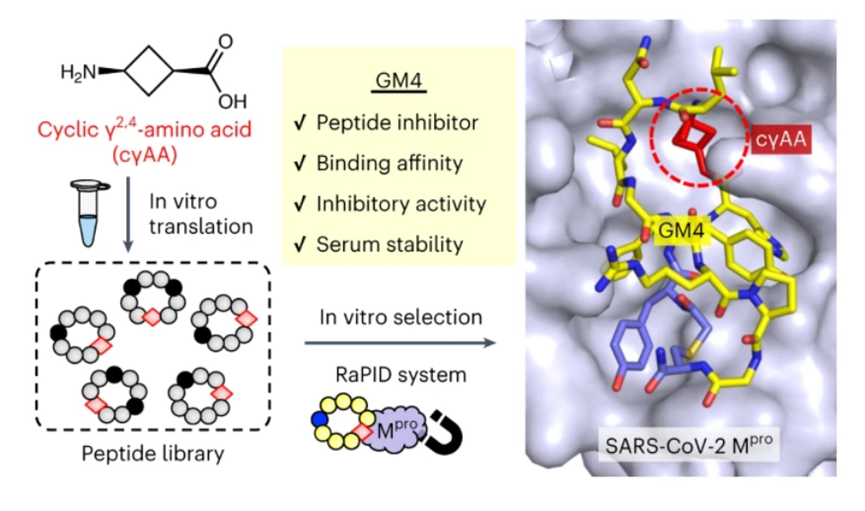
最后,作者通过解析GM4和Mpro的X-ray晶体结构,发现GM4保守基序yFHγ1的四个残基分别占据占据着Mpro的活性位点的S4、S2、S1、S1’底物结合区域,从而与Mpro紧密结合,最为重要的是,环状γ2, 4氨基酸残基结构中的环丁烷以接近平面的构象占据着P1’位置,不仅有效的抑制了Mpro的活性,同时抑制了Mpro对GM4的水解作用 (图4)。
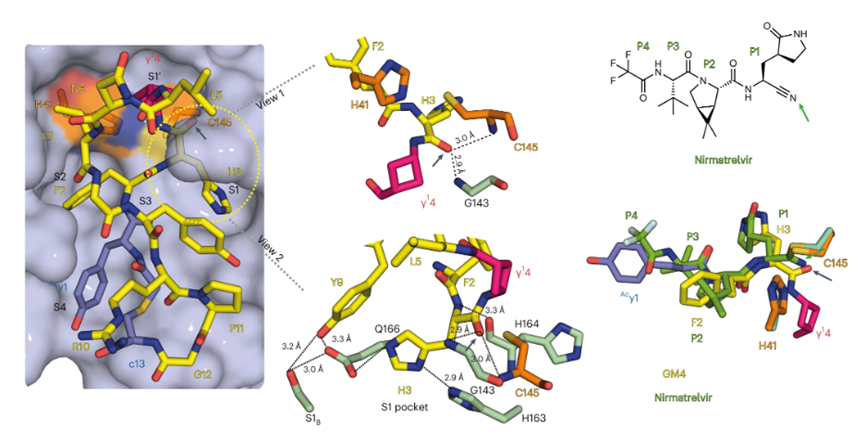
图4: GM4和Mpro的结合模式图
总结
总之,作者通过构象限制的环状γ2, 4氨基酸作为核糖体的延伸单元,证明含cγAAs环肽库构建的可行性。随后,作者通过随机非标准集成发现RaPID系统针对SARS-CoV-2主要蛋白酶筛选含有cγAAs的大环肽。并依据系统回收率选取7个含有cγAAs的大环肽 (GM1-GM7) 进一步分析其结合亲合力、抑制活性和蛋白水解稳定性。并将cγAAs突变成丙氨酸以验证cγAAs对于活性和水解稳定性的重要性。遗憾的是,由于GM4较差的膜渗透性导致其在100 μM下仍未表现出细胞毒性。后续作者设想通过在保持GM4关键的yFHγ1结合基序的同时降低分子量并增加化合物的膜渗透性。总之,cγAAs独特的折叠构象拓宽了大环的化学空间,包含cγAAs的核糖体合成的大环肽库结合RaPID系统一起构建了具有吸引力的多肽类新药研发平台。
参考文献
【1】 Miura T, Malla T R, Owen C D, Tumber A, Brewitz L, McDonough M A, Salah E, Terasaka N, Katoh T, Lukacik P, Damerell C S, Mikolajek H, Walsh M A, Kawamura A, Schfield C J, Suga H. In vitro selection of macrocyclic peptide inhibitors containing cyclic γ2, 4-amino acids targeting the SARS-CoV-2 main protease. Nat. Chem., 2023, 15: 998-1005. DOI: 10.1038/s41557-023-01205-1.

