PNAS:磷脂酰肌醇4,5-二磷酸耗尽可能是脑小血管病的普遍特征
时间:2021-06-12 06:01:47 热度:37.1℃ 作者:网络
脑小血管疾病(Cerebral small vessel diseases, CSVDs)是一组病理疾病,占缺血性中风的25%以上,占所有痴呆的40%以上。虽然已经发现了遗传形式,但散发性SVDs最为常见,并且发病率随着年龄的增长而增加。散发性SVD的原因仍然不清楚,目前没有治疗方案。SVDs可以发生在身体的任何器官。然而,大脑的微血管系统特别容易出现功能障碍。
最近PNAS杂志撰文探讨了磷脂酰肌醇4,5-二磷酸(PIP2)在CSVDs的神经血管藕合受损中的重要作用。

骨骼肌的新陈代谢需求部分是通过器官范围内的血管扩张来满足的,这种扩张降低了血液流动的阻力,因此增加的需求是通过组织内的血液流动来满足的。相比之下,颅骨的体积基本上是固定的,以防止大脑中血量的整体增加。因此,自然界进化出了独特的脑循环机制,以其它部位血流减少为代价,迅速将血液流向代谢活性较高的脑区。这一过程被称为功能性充血。它涉及到活跃的大脑区域和脑血管系统之间的通信,其过程被称为“神经血管耦合”。
在脑SVDs中,神经血管耦合被破坏,功能性充血的减弱状态导致血管认知障碍和痴呆。
Dabertrand等人在PNAS发文论证了一种特殊SVD功能性充血缺失的分子基础,并令人印象深刻地揭示了功能障碍是如何逆转的。特别是,外源性供应少量磷脂【磷脂酰肌醇4,5-二磷酸(PIP2)】可挽救大脑常染色体显性动脉病伴皮层下梗死和白质脑病(CADASIL)小鼠模型的受损的功能充血。这是主要的遗传性SVD和更常见的散发型SVD的模型。Dabertrand等人的发现可能为神经血管耦合损伤和大脑SVD相关痴呆治疗的发展奠定了基础。

大脑的脉管系统是有层次结构的组织。软脑膜动脉在皮层表面形成了一个高度互联的网络。不同分支直径的动态变化使软脑膜能够将血液输送到需要急性代谢的区域。这一网络穿透小动脉进入实质,进而提供一个巨大的、相互连接的毛细血管网络,为所有脑细胞提供能量底物。尽管血细胞从穿透性小动脉到最终通过穿透性小静脉再到静脉的过程中有大量的路径可走,但毛细血管为大脑中的血流提供了最大的阻力。
一个正在出现的大脑功能性充血模型集中于毛细血管。虽然不同脑区的毛细血管网络的密度是不同的,但从新皮层实质的一个位置到最近的毛细血管的标准距离很小,大约13 μm。Longden等人利用神经元和毛细血管之间这种隐性的、密切的关系,假设大脑使用毛细血管网络作为感觉网来检测神经元活动的升高,随后向上游穿透小动脉和血管发出扩张信号。其机制包括K+离子和内部整流的K+通道Kir2.1。每个神经元动作电位都会释放钾离子,原则上,在毛细血管附近的局部[K+]可以接近10mM。这个浓度足以激活Kir2.1,其开放阈值随着细胞外[K+]的增加而提高。这导致再生的超极化脉冲开始通过缝隙连接向邻近内皮细胞传播,从而进一步刺激Kir2.1通道活性以传播信号。在到达上游小动脉时,这种超极化信号通过缝隙连接传递到覆盖的平滑肌细胞,使它们舒张。小动脉平滑肌的舒张以及随后的穿透血管和小血管的扩张导致毛细血管网压力头的增加和通过活动增强区域的血流增加。最后,瞬时受体电位V4 (TRPV4)阳离子通道的激活以及可能的其他机制导致内皮细胞膜电位的恢复。
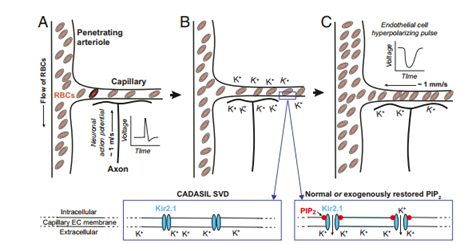
功能性充血的初始信号事件。(A和B)神经元动作电位通过细胞外[K+]的积累,在毛细血管内皮细胞中诱导超极化脉冲打开Kir2.1通道。这些通道的打开需要进一步结合PIP2。(C) K+通过开放的Kir2.1通道引起的毛细血管内皮细胞(ECs)的超极化脉冲,被电传导到上游穿透小动脉,并通过相关平滑肌的松弛诱导穿透小动脉的扩张。这导致毛细血管网络的压力差升高和红细胞流量增加。(插图)在CADASIL小血管疾病(SVD)模型中,PIP2的丢失阻止了Kir2.1通道的开放并阻断了功能性充血,而正常或恢复水平的PIP2支持Kir2.1通道的开放和正常的血流动力学反应。
基于K+/ kir2.1模型的探索使用了一种创新的体外大脑微血管制备,包括一个空心的、加压的贯穿小动脉段和一个完整的毛细血管树。直接刺激树与K+ bolus导致脑实质上游小动脉快速扩张。这种反应被低浓度的已知Kir2.1通道阻滞剂BaCl2阻断,而在内皮细胞特异性Kir2.1敲除小鼠中不存在。刺激触毛是神经血管生理学中一种经典的感觉通路,可引起触毛初级躯体感觉(vS1)皮层的功能性充血反应。在内皮细胞特异性kir2.1敲除小鼠中,注入BaCl2可减弱功能性充血,并使其受损。在后续研究中观察到PIP2在Kir2.1控制功能性充血机制中的强制性作用。特别是Harraz等人的研究显示,毛细血管内皮细胞中Gq G蛋白偶联受体通过磷脂酶C (PLC)发出信号,长时间刺激Gq G蛋白偶联受体,将削弱基于K+/ kir2.1的功能充血控制机制。有趣的是,其潜在机制是通过Gq信号通路消耗PIP2。
Dabertrand等证明,由于毛细血管内皮细胞Kir2.1通道的活性双重减弱,基于K+/Kir2.1的神经血管偶联机制在CADASIL SVD小鼠中缺失。这种缺陷导致vS1皮质中明显钝化触毛刺激诱导的功能性充血反应。
对PIP2水平降低的原因的调查最初集中在降低PIP2的过程上。特别是,传统认知指出PLC介导的水解PIP2下游的G qPCR信号。然而,PLC的抑制并没有影响Kir2.1电流活性的钝化,从而排除了降解途径。随后的研究探讨了脂质激酶合成PIP2,这是一种需要高ATP水平的低ATP亲和酶。值得注意的是,CADASIL SVD小鼠的脑毛细血管中ATP水平下降。这表明,脑毛细血管内皮细胞中ATP合成的减少是导致PIP2水平下降的最终原因,PIP2导致了级联反应,损害Kir2.1通道活性,导致CADASIL SVD小鼠神经血管偶联失败。
总之,Dabertrand等人表明,与CADASIL SVD相关的缺陷神经血管偶联的分子基础是一种由PIP2耗竭引起的毛细管Kir2.1通道病。这些发现为PIP2替代的治疗潜力提供了概念上的证明。更一般地说,它们能拼出大脑SVD的完整分子序列。然而,问题仍然存在。
PIP2耗竭依赖性微血管功能损害是CADASIL SVD模型特有的,还是会导致其他大脑SVD ?
最近的一份报告显示,PIP2治疗可恢复阿尔茨海默病5xFAD小鼠模型中受损的功能性充血,为广泛的以PIP2为中心的大脑微血管功能障碍模型提供了有趣的证据。作为一个潜在的临床问题,外源性供应的PIP2如何进入细胞?质膜上的翻转酶可以输入细胞外PIP2,但这一过程需要ATP,而CADASIL SVD小鼠的毛细血管受损。Ca2+依赖的混杂酶活性不依赖ATP,也可以将PIP2转运到质膜的叶。了解这种转运过程可能是打开PIP2补充治疗潜力的关键。
基于K+/ kir2.1的神经血管耦合机制似乎在1-s时间尺度上驱动了对大脑某一区域增加营养物质的最初反应。神经血管耦合的其它方面在更长的时间尺度上起作用。最近发现的一种互补机制是毛细血管内皮细胞中Ca2+内流通过瞬时受体电位固定蛋白1 (TRPA1)通道触发毛细血管到小动脉信号传导。由TRPA1启动的血管舒张信号在长时间的体感刺激中对维持功能性充血至关重要,它通过一种双相机制传播,涉及缓慢的细胞间Ca2+波以及Longden等人确定的kir2.1依赖的电传导机制。
因此,PIP2耗尽可能会损害大脑的功能性充血,而这种充血是由不同的分子传感器在多个时间尺度上协调的。PIP2缺失可能是大脑SVD的普遍特征。
原文出处
1、Earley S, Kleinfeld D. PIP2 as the "coin of the realm" for neurovascular coupling. Proc Natl Acad Sci U S A. 2021;118(21):e2106308118. doi:10.1073/pnas.2106308118
2、Dabertrand Fabrice,Harraz Osama F,Koide Masayo et al. PIP corrects cerebral blood flow deficits in small vessel disease by rescuing capillary Kir2.1 activity.[J] .Proc Natl Acad Sci U S A, 2021, 118: undefined.

