罕见病专栏|薄胼胝体和“猞猁耳征”——遗传性痉挛性截瘫11型一家系报告
时间:2025-01-19 12:13:42 热度:37.1℃ 作者:网络
摘 要 报道1例MRI影像表现为典型薄胼胝体(thin corpus callosum,TCC)和“猞猁耳征(ears-of-the-lynx sign)”的遗传性痉挛性截瘫11型(spastic paraplegia 11,SPG11)病例。患者为13岁男孩,主要表现走路不稳容易跌倒,症状逐渐加重伴双手书写不灵、学习能力下降1年;父母无血缘关系。患儿头颅MRI表现为胼胝体变薄,双侧脑室前角旁白质T2、Flair轴位对称性高信号,全外显子组测序检测到SPG11基因NM_025137:c.2073delT和c.257+5G>A两处杂合突变分别来自父母,先证者最终诊断为SPG11,并给予对症治疗。脑MRI影像表现TCC和“猞猁耳征”对诊断SPG11具有高度敏感性和特异性,对于有痉挛性截瘫表现的患者要考虑到SPG11的可能。
关键词
遗传性痉挛性截瘫11型;薄胼胝体;猞猁耳征;SPG11基因;复合杂合突变
遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP或称SPG)是一组具有高度临床和遗传异质性的单基因遗传性神经退行性疾病,单纯型HSP多为常染色体显性遗传,主要表现进展性双下肢无力和痉挛性截瘫,而复杂常染色体隐性遗传HSP(AR-HSP)亚型通常还存在智力减退、癫痫和特征性MRI等表现。SPG11是AR-HSP最常见的类型,由SPG11基因突变所致,MRI可表现为薄胼胝体(thin corpuscallosum,TCC)、脑萎缩和脑白质病变,“猞猁耳征(ears of the lynx sign)”是SPG11脑白质病变的一种表现。本文就1例具有典型TCC和“猞猁耳征”影像表现的SPG11报道如下。
1 临床资料
先证者(Ⅱ-1):男,13岁,2年前父母发现其走路不稳,跑步时易跌倒,症状逐渐明显,1年前发现右手书写不灵,学习能力下降,成绩较以前下降,注意力不集中,多动,无抽搐,无视觉、听觉和精神行为异常。发病以来患儿体质量无明显变化,饮食、睡眠及二便正常。患儿双眼近视,6岁时被牛顶伤右侧腹股沟部;其母孕期无特殊,生产时脐绕颈,无缺氧,正常喂养,否认家族史。
体格检查:体型偏胖,容貌正常,右侧颜面1处咖啡斑,右手示指、小指关节磨损样茧,双膝部新旧不同的伤疤,双踝关节轻度变形,双足内旋内翻。专科:神志清楚,精神正常,高级神经活动正常;脑神经检查正常;四肢肌容积、肌力和肌张力正常,双手轮替不协调,痉挛步态;深、浅感觉正常;双上肢腱反射正常,双侧膝腱反射亢进,双侧踝阵挛阳性,双侧病理征阳性;自主神经功能正常;眼科检查未见K-F环。
辅助检查:血浆氨、叶酸、Vit B12、铜蓝蛋白、血清铜、血乳酸、同型半胱氨酸正常;血清AQP-4、MOG、GFAP抗体阴性。超声检查提示右侧腹股沟局部皮下脂肪层回声增强(外伤有关)。脑电图、肌电图正常;双眼VEP:双眼P100潜伏期明显延长,波幅正常。小学文化程度,MMSE评分28(参考值30),MoCA评分28(参考值30)。颅脑MRI检查示胼胝体嘴、膝及前体部纤细,T2、Flair显示双侧脑室前角邻近白质对称性高信号。胸椎MR平扫和增强显示胸段脊髓略变细,见图1。
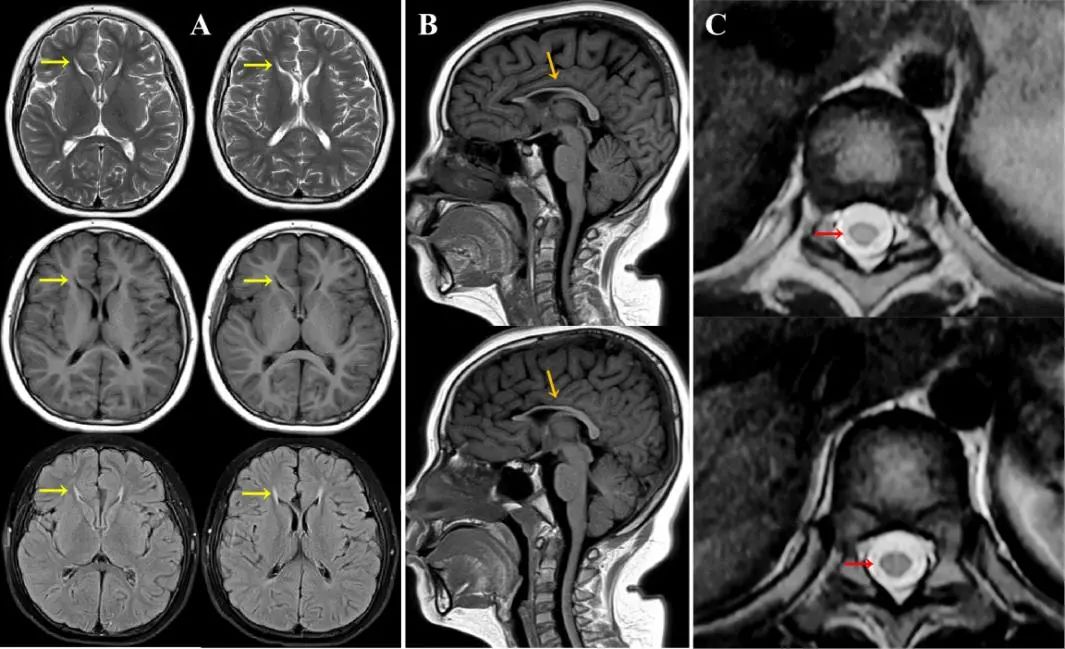
图1 头颅MRI表现 轴位(A),双侧脑室邻近前角白质T2、Flair高信号,T1低信号,呈“猞猁耳征”(黄箭头);矢状位(B),薄胼胝体(棕箭头);冠状位(C):胸髓轻度萎缩(红箭头)。Fig.1 MRI performance
基因检测:征得患儿及家属同意,采集EDTA抗凝外周血进行了全外显子组测序(whole-exome sequencing,WES)检测,发现患儿SPG11基因NM_025137:c.2073delT和c.257+5G>A两个位点杂合变异。为进一步遗传学分析,对患儿父母也进行了SPG11基因Sanger测序验证,父亲存在c.2073delT杂合变异,母亲存在c.257+5G>A杂合变异,患儿两处变异来自父母,呈复合杂合突变,支持AR遗传模式,见图2。c.2073delT为移码变异,会使终止密码子提前出现,导致截短蛋白质或无义介导的mRNA降解,MAZZONI等[1]认为提前终止编码的mRNA很不稳定,表达的蛋白也是异常的。该变异在正常人群数据库频率小于0.001(AR),根据ACMG《序列变异解读标准与指南》等相关证据,c.2073delT变异被判断为可能致病(LP)。另外一个c.257+5G>A变异位于经典剪接位点附近,通过MaxentScan、dbscSNV、splicePort软件预测,提示该位点可能影响剪接,对基因/基因产物有害或影响剪接的程度达到支持阈值水平,属于有害突变。根据ACMG《序列变异解读标准与指南》等相关证据,该变异被判断为意义不明(VUS)。
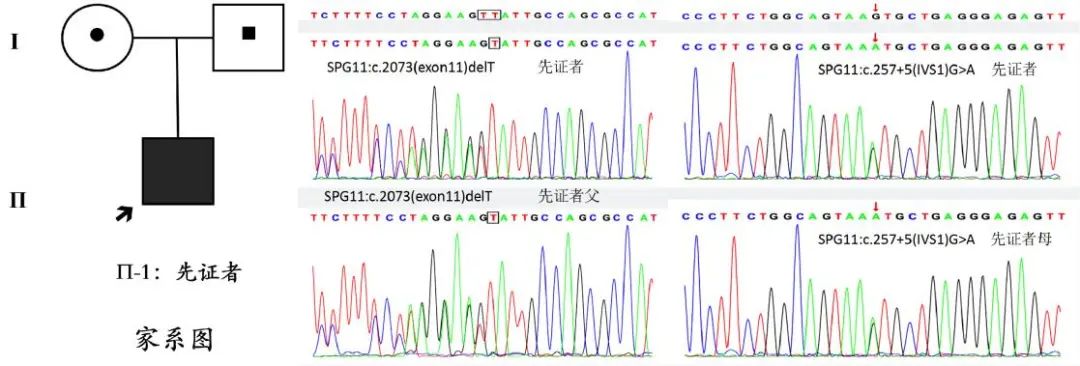
图2 家系图和基因突变分析 先证者SPG11基因c.2073delT和c.257+5G>A两个位点的变异分别来自父母,为复合杂合突变。Fig.2 Genetic pedigree diagram and genetic test results
根据临床表现和基因检测结果,该患儿最后诊断为SPG11,给予巴氯芬、胞磷胆碱口服,同时进行物理康复、股四头肌电刺激治疗,症状略有缓解,建议半年复诊一次。
2 讨论
遗传性痉挛性截瘫(HSP或称SPG)表型和基因型复杂多样[2],临床以双下肢痉挛性无力为核心症状。常染色体显性遗传的HSP(AD-HSP)占所有HSP的70%~80%,其中SPG4约占AD-HSP的40%。SPG11是最常见的AR-HSP,占所有AR-HSP患者的18.9%[3]。HSP伴薄胼胝体(hereditary spastic paraplegia with thin corpus callosum,HSP-TCC)在AR-HSP中很常见,其特征性影像表现是TCC,主要见于SPG11和SPG15[4]。SPG11临床表现复杂,与其他SPG亚型表型有重叠[5],除了有典型SPG的特点,还可有构音和吞咽障碍、智力发育迟缓或减退、视听觉异常、癫痫发作、共济失调、皮肤病变等表现[6],少数患者可伴有多巴反应性帕金森叠加综合征和周围神经病[7]。DU等[8]对339例SPG11患者临床特征、基因型与表型相关性研究发现SPG11具有明显的临床和遗传异质性,表型与基因型无明确相关性。该患儿临床表现为双下肢痉挛性无力,伴有智力发育减退和亚临床视神经损害,仅凭症状诊断SPG有一定难度,需要与许多疾病如脊髓压迫症、多发性硬化、神经梅毒、脊髓亚急性联合变性以及ALS、SCA、Krabbe病等[9]进行鉴别。
SPG11由SPG11基因缺陷所致,机制复杂,目前仍不完全清楚。SPG11的病理表现以胸段脊髓运动神经元轴索变性和神经元丢失为主,可伴有脱髓鞘改变[10],外侧膝状体、下丘脑变性是特征性病变部位,其神经细胞存在p62反应性包涵体和α-突触核蛋白相关的路易小体[11],这些变化使得SPG11的临床表现更加复杂,因此确诊SPG11仍有赖于基因检测。SPG11基因位于15q13-15,2007年被克隆,已确认的突变位点有180多个[12],国内已报道致病突变位点有近40个,多为移码突变[6]。SPG11基因编码的Spatacsin是一种潜在的跨膜蛋白,在小脑、大脑皮质、海马和松果体等处神经细胞核周及膜上有广泛表达,轴突、树突以及突触连接处更显著。Spatacsin可能通过基因调控、蛋白修饰、溶酶体-自噬及细胞分裂参与维持细胞骨架稳定性、调控突触小泡转运与可塑性等功能,Spatacsin异常会造成细胞功能障碍及髓鞘形成不良而致病[13]。最近有研究认为异常Spatacsin神经毒性也是疾病发生发展的机制之一,遗憾的是Spatacsin具体定位与功能目前仍不明确[11,13]。
该患儿WES检测和家系共分离验证发现SPG11基因存在c.2073delT和c.257+5G>A变异,这两处变异分别来自父亲和母亲。文献复习未发现这两个位点变异的SPG11个案或家系报道,可能为新发变异位点,有关人群携带率和突变对基因序列和蛋白质结构的影响尚不清楚。SPG11基因变异关联的疾病为SPG11、腓骨肌萎缩症轴索型X型(CMT2X)和青少年型肌萎缩侧索硬化5型(ALS5)。CMT2X是轴索损害为主的周围神经病,极罕见,临床以四肢远端进行性肌无力、肌萎缩和腱反射减退或消失为主要表现,神经传导速度减慢,与该患儿不符,可以排除;青少年型ALS5肌萎缩明显,肌电图有广泛神经源性损害可资鉴别。综合分析,SPG11基因变异符合复合杂合突变,具有致病性,结合患儿临床表现及典型影像特点并排除CMT2X和青少年型ALS5,确诊为SPG11(OMIM:604360)。该患儿SPG11基因两个变异位点目前尚未见有报道,其表型与已经报道的其他变异位点的表型并无不同。来自母亲的c.257+5G>A变异不是经典剪切突变位点,有进一步验证变异对基因功能影响的必要。
“猞猁耳征”又称“山猫耳征”,是脑白质病变的一种表现形式,指邻近侧室前角白质在T2和Flair成像呈现对称高信号,因轴位形似猞猁耳尖簇状毛而得名。TCC和“猞猁耳征”对诊断SPG11和SPG15的敏感性和特异性高[14],研究发现60%的AR-HSP患者是HSP-TCC,其中41%~77%是SPG11和SPG15,因此有TCC表现的痉挛性截瘫患者,即便没有家族史也要考虑AR-HSP可能[15]。约有5%的AR-HSP患者有“猞猁耳征”,仍以SPG11、SPG15多见,但研究发现同时具有TCC和“猞猁耳征”的SPG11患者并不多见[16]。除了SPG11和SPG15,部分Marchiafava-Bignami病、遗传性脊髓小脑性共济失调等疾病也有类似TCC和“猞猁耳征”的影像表现,可单独或同时出现,需要注意鉴别。该患儿临床考虑SPG11并确诊主要是基于影像学线索,因此,临床实践中重视和强调神经影像在SPG11诊断中的作用是恰当的。
SPGs目前无有效治疗方法,口服巴氯芬、替扎尼定等可缓解肌痉挛症状,口服无效或运动严重受损的患者可注射肉毒毒素或鞘内注射巴氯芬;有尿急的患者可用奥昔布宁、索利那新和抗胆碱能药物治疗[17]。针对单个SPG亚型治疗的研究如靶向分子疗法和个体化药物治疗取得了一些进展[18],有研究发现SPG11患者中存在较高的肥胖率,而肥胖与SPG11下丘脑神经变性之间存在联系,推测通过特定代谢干预控制肥胖可能延缓SPG11的运动受损[19]。对于运动功能严重受损的SPG患者需要多学科联合全程化病情评估和管理,多数SPG11患者发病后10~20年需要坐轮椅,死因多为并发症如肺炎等。预防SPG主要是针对患者家系的筛查、遗传咨询和婚育指导。
3 点评
SPG11是AR-HSP中最常见的类型,具有高度的临床和遗传异质性,致病基因SPG11基因突变谱广且复杂,以移码突变最常见。影像学TCC和“猞猁耳征”对诊断SPG11敏感性和特异性均高,对有上述征象的痉挛性截瘫患者要考虑SPG11和SPG15可能,确诊和分型需要基因检测。SPG11患者需要多学科全程化管理,有效治疗取决于靶向分子治疗的研究进展。
参考文献:
1. MAZZONI C, FALCONE C. mRNA stability and control of cell proliferation[J]. Biochem Soc Trans, 2011, 39(5): 1461-1465.
2. HEDERA P, ADAM M P, FELDMAN J, et al. Hereditary Spastic Paraplegia Overview[J]. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
3. FINK J K. Hereditary spastic paraplegia[J]. Curr Neurol Neurosci Rep, 2006, 6(1): 65-76.
4. 李爽, 刘小民. 遗传性痉挛性截瘫伴胼胝体发育不良的遗传学研究进展[J]. 国际神经病学神经外科学杂志, 2020, 47(5): 555-558.
5. PENSATO V, CASTELLOTTI B, GELLERA C, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15,SPG35 and SPG48[J]. Brain, 2014, 137(Pt 7): 1907-1920.
6. DU J. Research on clinical and molecular genetics of hereditary spastic paraplegia 11 patients in China[J]. Zhong Nan Da Xue Xue Bao Yi Xue Ban, 2022, 47(12): 1729-1732.
7. HARDING AE. Classification of the hereditary ataxias and paraplegias[J]. Lancet, 1983, 1(8334): 1151-1155.
8. DU J. Hereditary spastic paraplegia type 11: Clinicogenetic lessons from 339 patients[J]. J Clin Neurosci, 2021, 85: 67-71.
9. 姚莉, 田沃土, 曹立. 遗传性痉挛性截瘫诊断策略[J].中国神经精神疾病杂志, 2023, 49(2): 112-119.
10. MEYYAZHAGAN A, ORLACCHIO A. Hereditary Spastic Paraplegia: An Update[J]. Int J Mol Sci, 2022, 23(3): 1697.
11. HAYAKAWA M, MATSUBARA T, MOCHIZUKI Y, et al. An autopsied case report of spastic paraplegia with thin corpus callosum carrying a novel mutation in the SPG11 gene: widespread degeneration with eosinophilic inclusions[J]. BMC Neurol, 2022,22(1): 2.
12. BOUTRY M, MORAIS S, STEVANIN G. Update on the Genetics of Spastic Paraplegias[J]. Curr Neurol Neurosci Rep, 2019, 19(4): 18.
13. REGENSBURGER M, KRUMM L, SCHMIDT M A, et al. Neurometabolic Dysfunction in SPG11 Hereditary Spastic Paraplegia[J]. Nutrients, 2022, 14(22): 4803.
14. PATEL S, SETHI P K, ANAND I, et al. Hereditary spastic paraplegia with a thin corpus callosum due to SPG11 mutation[J]. Neurol India, 2016, 64(1): 171-172.
15. BOUKHRIS A, STEVANIN G, FEKI I, et al. Hereditary spastic paraplegia with mental impairment and thin corpus callosum in Tunisia: SPG11, SPG15, and further genetic heterogeneity[J]. Arch Neurol, 2008, 65(3): 393-402.
16. PASCUAL B, DE BOT ST, DANIELS MR, et al. “Ears of the Lynx” MRI Sign Is Associated with SPG11 and SPG15 Hereditary Spastic Paraplegia[J]. AJNR Am J Neuroradiol, 2019, 40(1): 199-203.
17. 何秉涛, 黄美欢, 贠国俊, 等. 遗传性痉挛性截瘫基因分型、分子诊断和治疗的研究进展[J]. 中国优生与遗传杂志, 2022, 30(3): 516-519.
18. SHRIBMAN S, REID E, CROSBY A H, et al. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches[J]. Lancet Neurol, 2019, 18(12): 1136-1146.
19. CARDOZO-HERNANDEZ A L C, REZENDE T J R, FRANCA M C J R. Hereditary spastic paraplegia type 11(SPG11) is associated with obesity and hypothalamic damage[J]. J Neurol Sci, 2020, 416: 116982.
【引用格式】于宗泳,武紫阳,田霏霏,等 . 薄胼胝体和“猞猁耳征”——遗传性痉挛性截瘫11型一家系报告[J]. 中国神经精神疾病杂志,2024,50(10):632-635.
【Cite this article】YU Z Y,WU Z Y,TIAN F F,et al.Thin corpus callosum and “lynx ear sign”: A report of a family of hereditary spastic paraplegia type 11[J]. Chin J Nervous Mental Dis,2024,50(10):632-635.
DOI:10.3969/j.issn.1002-0152.2024.010.012

