【论肿道麻】《Nature Metabolism》:葡萄糖限制保护癌细胞免于嘧啶限制诱导的凋亡
时间:2025-01-17 12:09:04 热度:37.1℃ 作者:网络
肿瘤细胞因其旺盛的增殖能力以及相对不完善的血供系统,常常处于营养匮乏的环境中,但令人意想不到的是极为有限的生存资源反而成为了它们进化出更强适应力的温床。做为细胞的核心碳源,葡萄糖不仅为细胞提供能量,还参与DNA、RNA和磷脂的合成,同时支持氧化还原活性分子的生成。在肿瘤微环境中,葡萄糖的含量极度匮乏,这一现象暗示着癌细胞面临着代谢进化的压力。
2024年Minwoo Nam等人在《Nature Metabolism》上发表的一篇研究,通过CRISPR筛选,探讨了葡萄糖限制在保护癌细胞免受嘧啶限制和复制抑制诱导的凋亡中的作用。研究发现,与肿瘤相关的低葡萄糖浓度能够保护肿瘤细胞免受嘧啶合成抑制的影响,这一生物合成途径是化疗中常见的靶点。研究发现了两种机制来解释这一在多种癌细胞类型中广泛观察到的现象。首先,低葡萄糖浓度限制了尿苷-5-二磷酸-葡萄糖(UDP-葡萄糖)的合成,从而保留了嘧啶核苷酸的可用性,并因此延长了复制叉停滞的时间。其次,低葡萄糖浓度通过抑制BAK的激活以及随后的细胞色素c的释放直接调节复制叉停滞下游的凋亡信号通路。这些结果表明,肿瘤中常见的低葡萄糖浓度可能会限制特定化疗药物的疗效,这一发现突显了考虑肿瘤营养环境对肿瘤治疗效果影响的重要性。对于实体瘤和血液瘤而言,肿瘤营养物质的可用性可能会受到肿瘤细胞、浸润的免疫细胞和非转化的基质细胞之间相互作用的影响。由于独特的代谢适应性可能支持癌细胞的增殖和存活,这为选择性地靶向癌细胞提供了机会,因此,增进我们对肿瘤营养环境对癌细胞代谢和信号传导途径影响的理解,对于改善肿瘤治疗是必要的。
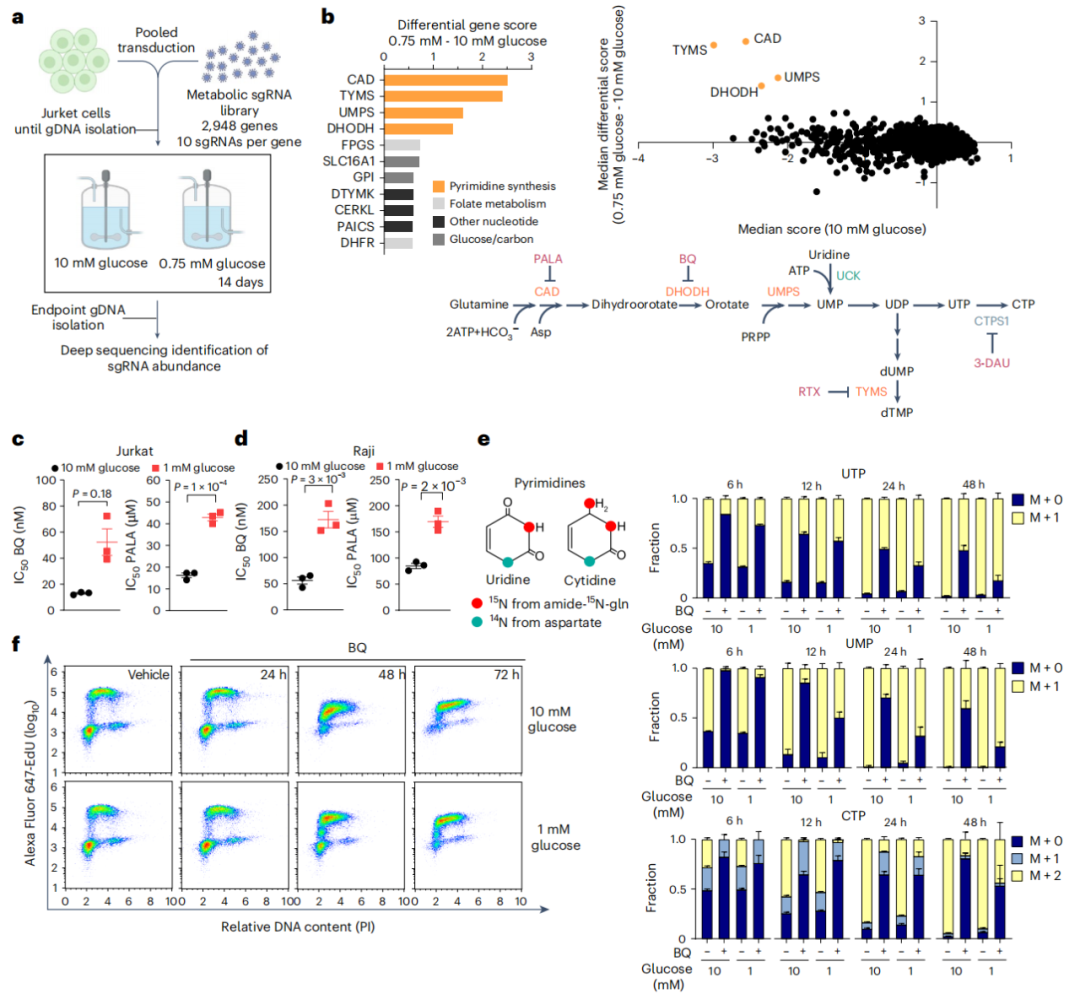
图1.低葡萄糖环境防止肿瘤细胞的嘧啶从头合成途径被抑制
嘧啶生物合成的抑制呈现葡萄糖依赖性特征
在肿瘤细胞中,本研究实施了一项针对3000个代谢基因的CRISPR筛选,这些基因编码了一整套代谢酶和转运蛋白。筛选在高葡萄糖(10 mM)以及与肿瘤相关的低葡萄糖条件(0.75-1 mM,以下简称低葡萄糖;图1a)下进行。本研究选用Jurkat细胞作为实验模型,原因在于其悬浮生长特性允许在连续流动细胞培养装置(称为Nutrostat)中进行培养,并且在这些条件下,Jurkat细胞对低葡萄糖浓度仅有轻微的敏感性。先前利用该数据集的研究集中于在低葡萄糖条件下选择性必需的基因,其中几乎所有的基因均支持线粒体功能。在本项研究中,研究焦点集中于那些在低葡萄糖条件下抑制效果得到缓解的基因,与标准条件相比,依据它们的差异基因得分对基因进行排序(图1b)。排名前四的基因——CAD、DHODH、UMPS和TYMS——编码的酶属于控制嘧啶从头合成的单一代谢途径(图1b)。为了探究这一结果是否是由于葡萄糖限制对代谢基因抑制敏感性的普遍影响,本研究将标准培养条件下代谢基因抑制的效果与低葡萄糖条件下的差异效果进行了比较,未发现普遍的相关性(图1b)。因此,葡萄糖限制对嘧啶合成需求的影响具有高度特异性。鉴于Jurkat细胞在Nutrostat中的高葡萄糖和低葡萄糖条件下几乎以相同的速率增殖,且通过从头途径的核苷酸扩张对于增殖细胞至关重要,这些结果出乎意料。本研究使用嘧啶生物合成酶的抑制剂来验证这些结果(图1b)。N-磷酰乙酰-L-天冬氨酸(PALA)抑制了氨基甲酰磷酸合成酶2、天冬氨酸转氨甲酰酶和二氢乳清酸酶(CAD)的天冬氨酸转氨甲酰酶活性,CAD是一种催化嘧啶从头合成前三个步骤的多功能酶;而Brequinar(BQ)抑制了二氢乳清酸脱氢酶(DHODH),这是该途径中的第二个酶。鉴于核苷酸耗竭会导致DNA复制叉停滞,进而导致S期延长或细胞周期停滞,本研究通过检测细胞增殖和5-乙炔基-2'-脱氧尿苷(EdU)基础的细胞周期分析来评估低葡萄糖条件下途径抑制的相对敏感性。与筛选结果一致,BQ和PALA的半数抑制浓度(IC50)在低葡萄糖条件下分别增加了四倍和2.6倍(图1c)。本研究在Raji细胞中也得到了类似的结果,Raji细胞是一种源自Burkitt淋巴瘤患者的B淋巴细胞系,其增殖不受低葡萄糖培养的影响(图1d)。鉴于嘧啶补救途径在CAD和DHODH的下游运作(图1b),尿苷可以通过尿苷-胞嘧啶激酶为嘧啶生物合成途径提供原料,从而绕过它们的抑制。尿苷挽救了BQ和PALA的抑制效应,表明这些小分子对增殖的影响是由于对嘧啶合成的靶向抑制。
随后,本研究进一步探讨了葡萄糖限制是否能够在抑制嘧啶合成的情况下,依然维持整体的嘧啶池。然而,本研究未能检测到显著的保护作用。本研究推测,在抑制嘧啶从头合成的条件下,核苷酸的消耗超过了其需求;因此,任何产生的嘧啶几乎立即被利用。因此,检测嘧啶池极为困难,而嘧啶的可用性可能仅通过DNA大分子的持续合成来体现。为了验证这一假设,本研究利用谷氨酰胺追踪技术来查询在葡萄糖限制和DHODH抑制下嘧啶的周转率。本研究观察到,在低葡萄糖条件下,包括尿苷单磷酸(UMP)、UTP和胞苷三磷酸(CTP)在内的嘧啶被标记的速度显著加快(图1e)。相比之下,包括肌苷单磷酸(IMP)(ATP和GTP的前体)、ATP和GTP在内的嘌呤池在低葡萄糖条件下的标记速度相似或仅略有增加。接着,本研究在单细胞和单个复制叉水平上监测了这些条件下的DNA合成。EdU掺入显示,在BQ处理后48小时内,细胞在S期停滞,而在低葡萄糖条件下这种停滞被延迟(图1f,g)。EdU掺入的定量分析还显示,在低葡萄糖条件下,经BQ处理的细胞具有更高的掺入率,表明葡萄糖限制支持DNA合成,尽管存在DHODH抑制(图1h)。为了更好地理解嘧啶耗竭对复制叉动态的影响,本研究进行了DNA纤维分析,该分析可测量单个复制叉上核苷酸的掺入速率。正如预期的那样,DHODH抑制降低了复制叉的速度并增加了复制叉的停滞(图1i,j)。重要的是,低葡萄糖培养显著减轻了DHODH抑制的这些效应,而单独的低葡萄糖培养并不影响复制叉动态和细胞周期分布(图1i,j)。在Raji细胞中也观察到了类似的结果。这些数据表明,在嘧啶耗竭的情况下,葡萄糖限制允许在单个复制叉水平上继续进行核苷酸掺入。
随后,本研究进一步探究了低葡萄糖培养条件下的增殖优势是否同样适用于嘌呤合成的抑制。为解答此问题,本研究采用了两种不同的IMP脱氢酶抑制剂——米多滨和麦考酚酸(MPA),以阻断GMP的生成。与BQ和PALA不同,低葡萄糖条件仅对这些IMP脱氢酶抑制剂的半数抑制浓度(IC50)产生边缘性影响。此外,氨甲喋呤(一种二氢叶酸还原酶抑制剂,能够阻断嘧啶和嘌呤的从头合成)的IC50值未发生改变,这表明葡萄糖限制对嘧啶合成抑制具有选择性效应,与本研究的筛选结果相一致。此种差异可能归因于嘌呤限制对众多细胞基本功能的影响,这些功能需要ATP和GTP的水解,预期会独立于复制或转录抑制而限制细胞的活性。这些结果进一步证实,葡萄糖限制的影响并非仅仅与因增殖差异而导致的核苷酸利用变化相关。因此,本研究得出结论,葡萄糖限制在嘧啶从头合成受到抑制时,特异性地促进肿瘤细胞的增殖。
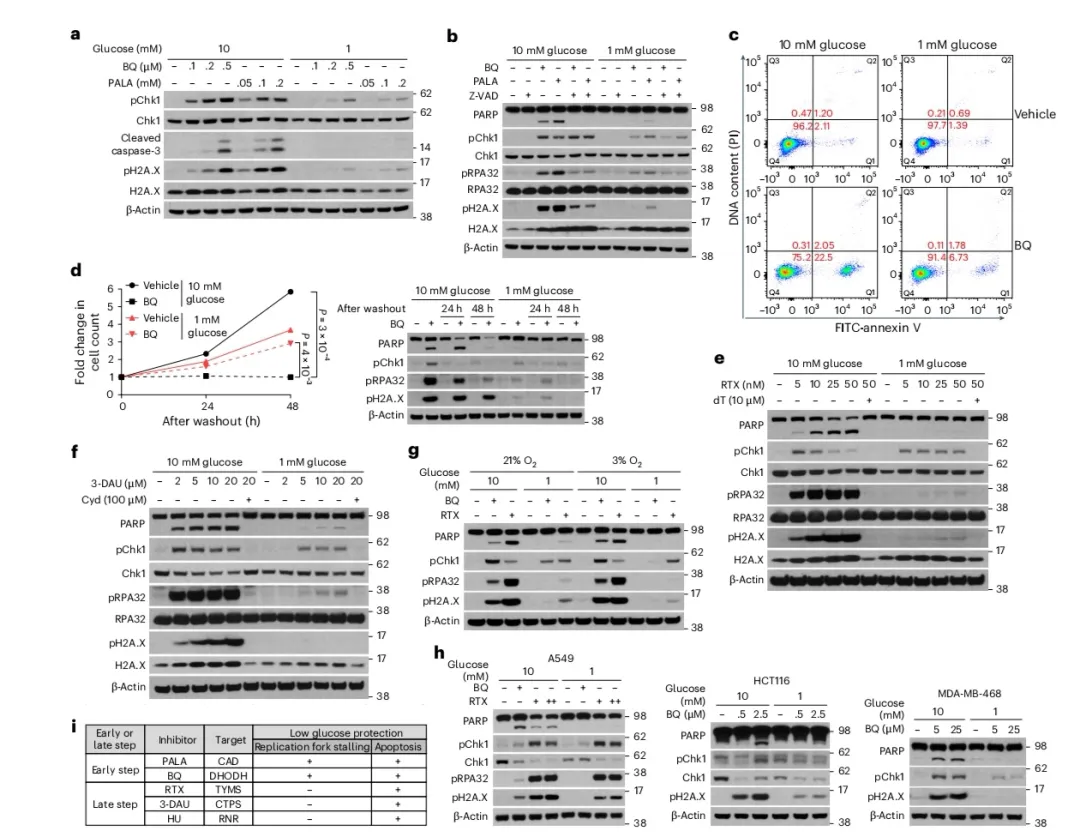
图2
血糖浓度对细胞复制及凋亡信号通路具有影响
复制叉停滞是复制应激的标志性特征。在停滞的复制叉处,ATR-Chk1信号通路被激活,以协调多个下游过程,这些过程有助于稳定复制叉并修复受损的DNA。本研究旨在探究由BQ或PALA处理所诱导的嘧啶核苷酸耗竭是否引发复制应激和凋亡,以及低葡萄糖是否具有保护作用。在抑制CAD或DHODH后,磷酸化Chk1(Ser345),作为复制叉停滞的标志物,其表达水平上升(图2a)。此外,经BQ和PALA处理的细胞中,通过磷酸化pA.X(Ser139)评估的DNA双链断裂、通过磷酸化RPA32(Ser4/Ser8)评估的单链DNA以及通过caspase-3和聚(ADP-核糖)聚合酶(PARP)裂解评估的凋亡均被诱导(图2a、b)。值得注意的是,低葡萄糖条件显著抑制了上述所有反应(图2a、b),而尿苷补充则可恢复这些反应。泛caspase抑制剂Z-VAD-FMK(Z-VAD)能够逆转BQ诱导或PALA诱导的PARP裂解,而不影响磷酸化Chk1的水平,这表明凋亡是在复制叉停滞的下游被激活的(图2b)。磷酸化RPA32(Ser4/Ser8)和磷酸化pA.X主要通过Z-VAD恢复(图2b),这表明在阻断嘧啶从头合成时,这些DNA损伤的后果主要是在凋亡执行过程中被诱导的,正如先前在由基因毒素和FAS信号诱导凋亡的细胞中所报道的那样。本研究采用annexin V和碘化丙啶(PI)染色,定量评估单细胞的凋亡情况。在BQ处理后72小时,低葡萄糖条件下凋亡细胞(annexin V+/PI−)的比例显著降低(7%对比23%;图2c)。随后,通过洗脱药物并持续培养细胞长达48小时,以评估这些细胞的存活能力。结果显示,在高葡萄糖水平下,经BQ处理的细胞增殖停止,而在低葡萄糖水平下培养的细胞在药物移除后仍可继续增殖(图2d)。相应地,在高葡萄糖水平下,PARP裂解持续存在,细胞在洗脱后48小时内进入凋亡的晚期阶段,这通过完整和裂解的PARP水平的显著下降得以证实,而在低葡萄糖水平下未观察到凋亡标志物(图2d)。这些数据表明,低葡萄糖水平可显著抑制因嘧啶合成抑制而引发的凋亡过程。
BQ与PALA作用于嘧啶从头合成途径的初始上游步骤,阻断了包括核糖核苷酸及脱氧核糖核苷酸在内的所有嘧啶核苷酸的生成。鉴于此,本研究进一步探究了针对生成胸苷或胞嘧啶核苷酸的下游酶的抑制剂是否能够复现BQ与PALA诱导复制应激及凋亡的效果,以及低葡萄糖环境是否具有相应的保护作用。Raltitrexed(RTX)与3-deazauridine(3-DAU)分别靶向胸苷酸合成酶(TYMS)与胞苷三磷酸合成酶1(CTPS1)(图1b)。Hydroxyurea(HU)则抑制核糖核苷酸还原酶,该酶催化核糖核苷酸向脱氧核糖核苷酸的转化,进而抑制DNA复制。与BQ和PALA类似,这三种下游酶抑制剂(RTX、3-DAU、HU)在高葡萄糖条件下均能激活细胞内的复制应激与凋亡,这一结论通过检测pChk1及裂解的PARP得以验证(图2e、f)。低葡萄糖环境可有效抑制由这些下游酶抑制剂所诱导的凋亡(图2e、f)。对于RTX与3-DAU而言,其诱导的效应可通过相应的下游产物(胸苷或胞嘧啶)进行逆转,从而证实了在所使用浓度下抑制剂的靶向特异性(图2e、f)。先前的研究已表明,嘧啶失衡可导致复制叉停滞,而抑制这些针对单一嘧啶种类的晚期步骤酶预计会引发嘧啶核苷酸失衡。然而,本研究并未观察到通过联合使用3-DAU对RTX诱导的复制应激与凋亡产生显著的缓解作用,这表明嘧啶失衡并非复制应激的主要诱因。在洗脱实验中,无论葡萄糖浓度如何,RTX处理的细胞在无药物培养基中的增殖均被终止,这可能与RTX进入细胞后长时间滞留有关。因此,本研究在RTX洗脱时补充胸苷,并观察到在低葡萄糖条件下经RTX处理的细胞仍保留增殖能力,而在高葡萄糖条件下则未观察到此现象。此外,凋亡标志物仅在低葡萄糖条件下经RTX处理的细胞中得以清除。低葡萄糖的保护效应同样在模拟生理条件的培养基和氧浓度环境中得到体现(图2g)。相比之下,与该抑制剂的IC50值一致,嘌呤合成抑制剂MPA在高葡萄糖和低葡萄糖水平下均以剂量依赖性方式同等程度地诱导复制应激与凋亡。抑制嘧啶合成还可能限制rRNA和mRNA合成所需的核糖核苷酸,进而触发凋亡。然而,低葡萄糖对由放线菌素D(一种mRNA和rRNA合成抑制剂)处理诱导的复制应激与凋亡标志物并无影响,这表明改变的RNA合成并非抑制嘧啶合成触发凋亡的近端葡萄糖调节机制。
为将本研究发现拓展至源自实体瘤的贴壁癌细胞系,本研究对A549(KRAS突变型)、HCT116(KRAS突变型)及MDA-MB-468(PTEN缺失型)细胞系在嘧啶合成抑制条件下的反应进行了考察。基于先前研究发现KRAS突变型驱动的癌细胞或PTEN突变/缺失型癌细胞对DHODH抑制表现出易感性,故选取了上述细胞系。在全部三种细胞系中,依据PARP裂解与磷酸化pA.X的减少,低葡萄糖水平下抑制嘧啶合成所诱导的凋亡均受到抑制(图2h)。克隆形成实验亦支持在低葡萄糖水平下培养的经RTX处理的HCT116细胞存活率上升。在p53野生型的A549与HCT116细胞中,BQ处理导致Chk1降解以及Chk1磷酸化程度极低,从而无法对低葡萄糖对复制应激的抑制作用进行评估。与Jurkat细胞类似,表达突变且功能失常的p53的MDA-MB-468细胞在低葡萄糖水平下经BQ处理时亦抑制了Chk1磷酸化(图2h)。
接下来,本研究探讨了其他具有不同作用机制的DNA靶向剂诱导的凋亡是否亦可被低葡萄糖所抑制。尽管针对拓扑异构酶(依托泊苷与多柔比星)或造成DNA损伤(顺铂)的药物对Chk1磷酸化与凋亡的影响在低葡萄糖条件下未发生改变或仅受到轻微抑制,但复制性DNA聚合酶抑制剂阿糖胞苷诱导的凋亡在低葡萄糖条件下显著减弱。这些数据表明,当凋亡主要由复制抑制所诱导时,葡萄糖限制可抑制凋亡,而其他基因毒性应激主要通过一种与葡萄糖无关的不同机制触发凋亡。
综上所述,低葡萄糖水平会抑制嘧啶从头合成的多个步骤、脱氧核苷三磷酸的生成或复制阻断所诱导的凋亡。然而,值得注意的是,低葡萄糖对复制叉停滞(Chk1磷酸化)的影响因抑制嘧啶脱氧核苷酸合成的早期(CAD、DHODH)或晚期(TYMS、CTPS1、核糖核苷酸还原酶)步骤而异(图2i)。

图3
葡萄糖对嘧啶代谢的影响独立于嘧啶的补救合成途径
鉴于不同途径对嘧啶生物合成的影响可能在复制叉停滞方面存在差异,我们推测干扰这些途径的方式在受现有核糖核苷酸池影响方面有所不同。我们首先假设,葡萄糖限制可能通过尿苷促进嘧啶的补救合成,以规避早期步骤的抑制并缓解随后的复制应激。至少有两种可能的来源可以贡献于细胞内尿苷的可用性:(1)核糖体(核糖噬)的自噬降解,这是RNA的主要储存库;(2)血清尿苷,通常存在于1–10 μM的浓度。我们首先利用RPS3-Keima系统监测了在嘧啶从头合成抑制时的核糖噬。RPS3是40S核糖体的一个亚基,Keima是一种具有pH敏感的双重激发特性的荧光蛋白(中性与酸性条件下的对比)。Keima还对溶酶体蛋白酶具有抵抗力,这使得能够通过免疫印迹评估在经历核糖噬的细胞中RPS3-Keima的溶酶体处理。采用先前描述的CRISPR敲入方法,我们建立了表达RPS3-Keima-3×FLAG融合蛋白的Jurkat细胞,以替代两个内源性RPS3等位基因。我们通过用torin-1处理细胞来确认报告基因的功能,torin-1通过mTOR抑制诱导核糖噬,并观察到RPS3-Keima的处理。然而,在BQ和RTX处理下,在低葡萄糖条件下没有Keima处理的证据(图3a)。泛caspase抑制显示,观察到的少量处理可能是在凋亡下游发生的(图3a)。我们进一步测试了自噬回收核苷和核苷酸是否是低葡萄糖中Chk1磷酸化抑制的基础,通过敲除编码ATG7的基因,ATG7是自噬的一个关键因子。尽管ATG7的缺失阻止了自噬流,这通过成熟自噬体的标记物LC3B-II的消失得以证明,但我们观察到自噬抑制对BQ处理或RTX处理细胞的复制应激和凋亡没有显著影响(图3b),表明低葡萄糖的保护效应是自噬独立的。
接下来,本研究通过将细胞置于含常规血清(FBS)或透析FBS(dFBS)的培养基中培养,探究血清是否可作为重要的尿苷来源。经BQ处理后,与含FBS的培养基相比,含dFBS的培养基显著加速了Chk1磷酸化及凋亡进程(图3c),暗示血清尿苷对嘧啶核苷酸池的贡献不容忽视。相比之下,在经RTX处理的细胞中未观察到血清的差异性效应(图3c),RTX抑制了尿苷进入后的嘧啶从头合成途径。
因此,本研究通过敲除尿苷-胞嘧啶激酶(UCKs)来检验尿苷的补救是否是葡萄糖对嘧啶合成抑制效应的关键因素。UCK1和UCK2是磷酸化尿苷或胞嘧啶的关键酶,而UCK样1(UCKL1)是一种具有预测的UCK活性的同源酶。在初步尝试敲除UCK2时,未能成功获得UCK2缺失的克隆,这促使我们推测UCK2可能具有与其UCK活性无关的必需功能,正如其他研究者所提出的那样。实际上,在异位表达激酶失活的UCK2突变体(D62A)的细胞中,成功敲除了UCK2,并随后敲除了UCK1。这些双重敲除(KO)细胞保留了相当数量的UCK活性,这通过部分恢复嘧啶和Chk1磷酸化得以证实,从而确认了UCK活性的缺失(图3c)。在UCK三重敲除(tKO)细胞中未观察到FBS对复制应激和凋亡的保护效应(图3c),这进一步证实了血清尿苷可显著抑制嘧啶从头合成抑制的效应。
借助这些经过验证的UCK tKO细胞,本研究现可探究葡萄糖是否通过尿苷补救影响嘧啶的可用性。在BQ处理后24小时,UCK tKO细胞中Chk1磷酸化被显著诱导,这一效应仍可通过低葡萄糖培养得到抑制(图3d)。此外,低葡萄糖在UCK tKO细胞中抑制凋亡的效果与亲本细胞相当(图3d)。这些数据表明,尽管血清尿苷对嘧啶的可用性有显著贡献,但调节尿苷补救对于低葡萄糖保护细胞免受嘧啶合成抑制引起的复制叉停滞和凋亡并非必需。因此,本研究生成了一个同时缺乏预测的UCK,即UCKL1的细胞系(UCK三重敲除,tKO),并观察到尿苷无法挽救嘧啶从头合成抑制对细胞增殖的影响。
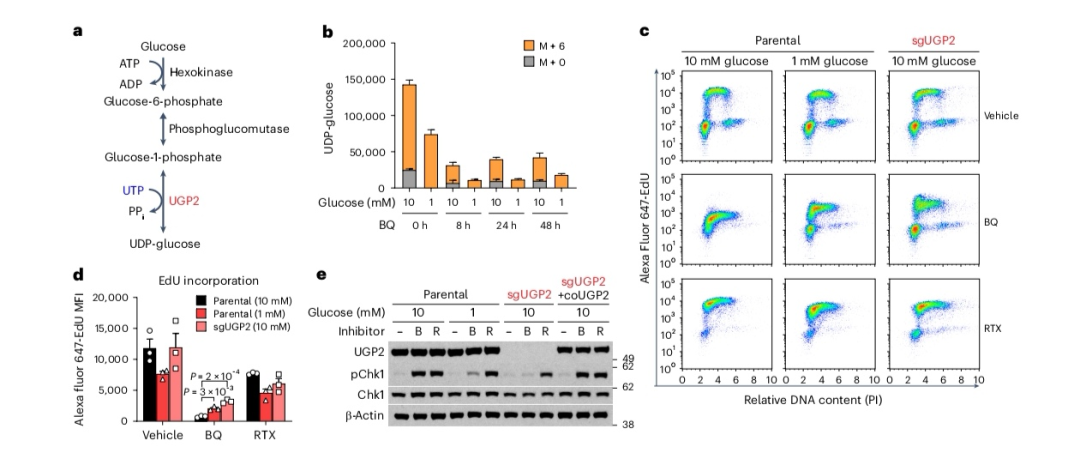
图4
葡萄糖促使UTP(尿苷三磷酸)转向UDP-葡萄糖(尿苷二磷酸葡萄糖)的合成过程
低葡萄糖条件下维持嘧啶可用性的另一种可能机制是限制嘧啶核苷酸在分支途径中的使用。嘧啶核苷酸糖是UTP的一个主要去向,UTP与葡萄糖-1-磷酸通过UDP-葡萄糖焦磷酸化酶2(UGP2)结合,形成UDP-葡萄糖(见图4a)。由于UGP2活性不受调控,且无机焦磷酸(PPi)能迅速被细胞内的无机焦磷酸酶代谢,正向反应占主导地位。在嘧啶受限条件下,UGP2和CTPS1可能竞争有限的UTP池。此外,人类CTPS1对UTP的相对较高的Km值(590 µM)与UGP2(76 µM)相比,表明UGP2介导的UDP-葡萄糖生成应主导于CTPS1介导的CTP生成。实际上,我们观察到在BQ处理下,UDP-葡萄糖水平的保持情况比CTP水平更好(剩余19%的UDP-葡萄糖,而CTP几乎无法检测到)。为了测试是否有相当数量的UTP可以被引导至UDP-葡萄糖合成,我们首先了解葡萄糖的可用性是否影响UDP-葡萄糖的合成。[U-13C]葡萄糖示踪显示,与高葡萄糖相比,低葡萄糖条件下既有(M + 0)又有新合成的(M + 6)UDP-葡萄糖水平均降低(图4b)。尽管BQ处理早在8 h就降低了UDP-葡萄糖水平,但高葡萄糖条件下的细胞保留了显著更多的UDP-葡萄糖(图4b)。
因此,我们考虑UGP2缺失是否会在高葡萄糖条件下保护细胞免受DHODH抑制的影响,类似于低葡萄糖培养。UGP2缺失的Jurkat细胞比亲本细胞增殖更慢,这一效应可以通过引入UGP2互补DNA(cDNA)来挽救。值得注意的是,UGP2缺失细胞在高葡萄糖条件下对BQ处理的响应中,保持了与低葡萄糖条件下亲本细胞相似程度的EdU掺入能力(图4c,d)。相比之下,UGP2缺失并未影响细胞在用下游抑制剂RTX处理时的EdU掺入(图4c,d)。相应地,BQ诱导的Chk1磷酸化在UGP2缺失的情况下被抑制,与低葡萄糖培养中的情况相当,而RTX诱导的Chk1磷酸化则几乎不受UGP2缺失的影响(图4e)。重新引入UGP2恢复了其缺失对复制应激的保护效应(图4e)。总体而言,这些数据表明低葡萄糖阻止UTP转向UDP-葡萄糖合成途径,保留足够的嘧啶以延长在上游酶DHODH和CAD抑制下的DNA掺入。相比之下,抑制嘧啶从头合成的晚期步骤——即UTP-UDP-葡萄糖分流下游的步骤——仍然限制了嘧啶脱氧核苷酸池,并导致复制叉停滞,无论葡萄糖水平如何。
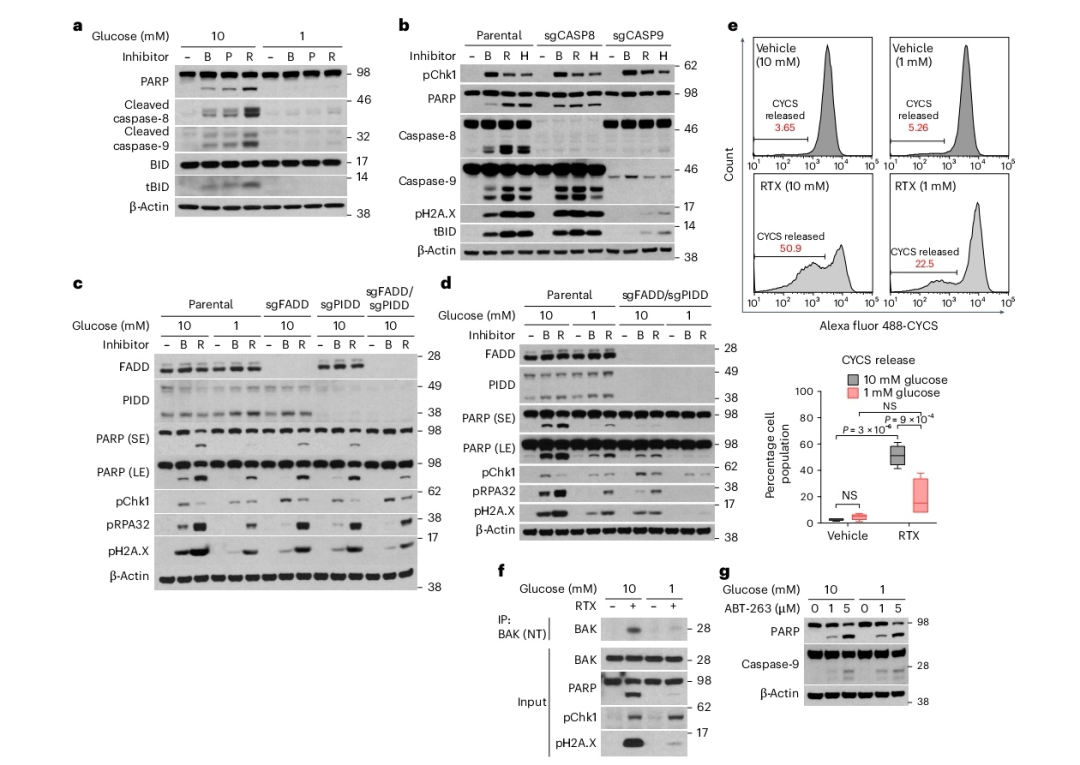
图5
低葡萄糖在复制抑制时减少凋亡
如上所述,葡萄糖影响复制叉停滞下游的第二个过程,该过程触发凋亡。基因毒性应激主要激活起始caspase,包括caspase-8和caspase-9,这些caspase随后激活执行caspase——caspase-3。嘧啶合成抑制剂促进了caspase-8和caspase-9的裂解,以及caspase-8的靶标BID的裂解(图5a)。尿苷和胸苷分别逆转了BQ或PALA和RTX处理时caspase-8和caspase-9的裂解,证实了caspase级联反应在嘧啶核苷酸耗竭诱导的复制应激下游起作用。这种起始caspase的激活在低葡萄糖条件下被阻断(图5a)。相比之下,低葡萄糖未能抑制FAS死亡受体介导的(caspase-8依赖的)和staurosporine诱导的(caspase-9依赖的)凋亡,表明低葡萄糖并非这些caspase级联反应的一般抑制剂。某些基因毒性应激因子(依托泊苷、多柔比星和紫外线)可以通过诱导FASL的分泌来诱导外源性FASL激活的FAS死亡受体信号传导。然而,用BQ或RTX培养的细胞的条件培养基未能在未暴露的细胞中诱导凋亡。在某些情况下,caspase-8(以及caspase-3和caspase-10)可以介导BID的截短,BID是一种促凋亡蛋白,可以激活MOMP,从而触发caspase-9的激活。因此,为了确定caspase-8和caspase-9在本系统中诱导凋亡的相对贡献,我们分别敲除了它们。值得注意的是,caspase-9的敲除完全阻断了BID的截短和嘧啶合成抑制时的凋亡诱导,而caspase-8是可有可无的(图5b)。我们还观察到,异位表达c-FLIP,它可以抑制caspase-8并可以完全阻断FAS介导的凋亡,尽管c-FLIP与procaspase-8和FADD的正常相互作用,但对BQ诱导或RTX诱导的凋亡没有影响。这些数据表明,caspase-9下游的caspase-3或caspase-8(或两者)的裂解启动了一个涉及BID的正反馈循环,这与先前的研究一致。鉴于caspase-3也介导caspase-8和caspase-10的裂解,我们得出结论,caspase-9和caspase-3下游的BID截短是通过这个放大循环实现的。
为了验证该放大循环在嘧啶合成抑制介导的凋亡中的必要性,本研究通过敲除FADD(一种含死亡结构域的接头蛋白,与caspase-8和caspase-10形成复合体并对其激活所必需)来探究其作用。在BQ或RTX处理下,FADD缺失细胞中的凋亡部分受到抑制,表明FADD复合体在caspase-9下游对凋亡有所贡献(图5c)。除FADD依赖途径外,复制应激下游还可激活一条PIDD驱动的途径。PIDD作为支架招募并激活procaspase-2,进而促进BID的截短。PIDD缺失在BQ或RTX处理时部分挽救了凋亡,与FADD缺失的效果相当,而FADD和PIDD的双重缺失则显著抑制了凋亡,表明FADD和PIDD共同对放大循环有所贡献(图5c)。然而,低葡萄糖培养的FADD或PIDD双重缺失细胞进一步抑制了凋亡(图5d)。综上所述,这些数据表明低葡萄糖调节caspase-9上游的凋亡信号,进而影响下游的caspase级联反应,包括caspase-3,并涉及FADD依赖和PIDD依赖的反馈放大循环,这些循环涉及caspase-8和BID。
鉴于caspase-9的激活是由MOMP介导的细胞色素c释放触发的,本研究考虑葡萄糖水平是否影响这些上游步骤。事实上,先前的研究表明线粒体状态可以影响凋亡的执行。本研究使用已建立的流式细胞术检测方法测量细胞色素c的释放,并观察到RTX处理后细胞色素c释放增加,这一效应在低葡萄糖条件下受到抑制(图5e)。相应地,4,6-二氨基-2-苯基吲哚(DAPI)染色显示低葡萄糖条件下DNA碎片化减少。
鉴于BAX和BAK构成细胞色素c释放的MOMP孔,且Jurkat细胞仅表达BAK,本研究生成了BAK敲除(KO)细胞系。观察到在BAK缺失时,嘧啶核苷酸耗竭和基因毒性应激下游的细胞色素c释放和凋亡被完全阻断。BAX和BAK的寡聚化及MOMP由Bp-only蛋白和Bp模拟物通过诱导BAX或BAK的构象变化触发。免疫共沉淀(IP)实验显示,在RTX处理时,低葡萄糖培养也阻断了由构象变化驱动的BAK激活(见图5f)。此外,Bp模拟物ABT-263和强力霉素(DOX)诱导表达的p15截短型BID(tBID),它们直接激活BAX和BAK,在高葡萄糖和低葡萄糖条件下均等程度地诱导caspase-9激活和凋亡(图5g)。如预期,这些刺激以BAK依赖的方式诱导凋亡。总体而言,这些结果表明葡萄糖特异性地调节起源于停滞复制叉下游核内的信号,该信号触发BAK激活、MOMP、caspase-9裂解和通过内源途径的凋亡。Caspase-8、BID及相关途径(p53诱导的含死亡结构域蛋白(PIDD)/FADD)可对凋亡激活程度有所贡献;然而,它们完全位于MOMP和caspase-9裂解的下游,并非caspase-3激活和PARP裂解所严格必需。尽管在核苷酸合成抑制时ATR–Chk1途径明确被激活,但该途径的激活是适应性的,并通过CDK2调控的S期检查点发生。因此,在高葡萄糖和低葡萄糖条件下,ATR或Chk1抑制均与嘧啶从头合成抑制协同作用,触发凋亡。
讨论与小结
以上发现不仅为癌症治疗提供了新的视角,也为未来的研究指明了方向。研究表明,肿瘤微环境中的低葡萄糖水平是如嘧啶合成抑制剂这类化疗药物疗效受限的潜在原因之一。这为优化抗癌治疗方案提供了新的科学依据,同时提示在开发新疗法时需充分考虑肿瘤微环境的代谢特性。
研究者们计划研究如何阻断其他癌细胞通路,从而引发对这些化疗的细胞凋亡。研究指出,已经存在一些实验药物,如Chk-1和ATR抑制剂,可能会实现这一目标,但需要更多的研究。此外,可以开发诊断测试来测量患者的癌细胞对低糖微环境最有可能做出的反应,并预测患者对特定化疗的反应。这些测试将有助于个性化医疗的发展,使治疗更加精准和有效。
低糖环境对肿瘤细胞的保护作用是一个复杂而引人入胜的领域,它不仅挑战了我们对肿瘤微环境的理解,也为癌症治疗提供了新的策略。随着研究的深入,未来有望开发出更有效的治疗方案,为患者带来新的希望。在与癌症的斗争中,每一次科学的进步都是我们宝贵的武器。

