病例报告|COQ8A基因突变所致原发性辅酶Q10缺乏症1例
时间:2025-01-16 12:11:07 热度:37.1℃ 作者:网络
摘 要 原发性辅酶Q10缺乏症属于常染色体隐性遗传病,由辅酶Q10生物合成过程中关键酶的编码基因发生突变导致。本文首次报道辅酶Q8A(coenzyme Q8A, COQ8A)基因一种新的杂合变异导致的原发性辅酶Q10缺乏症。患者为26岁男性,以缓慢起病的共济失调、运动不耐受、震颤以及癫痫发作为主要表现,诊断原发性辅酶Q10缺乏症,全外显子基因测序结果示COQ8A基因复合杂合突变[NM_020247.5:exon8:c.1009G>A(p.A337T)和NM_020247.5:exon8:c.1078delC(p.Q360Sfs*20)],予以补充辅酶Q10后症状有所改善。该病例提示当患者出现共济失调、运动障碍,尤其伴癫痫或认知功能障碍时,应排查可治性的原发性辅酶Q10缺乏症。
关键词
COQ8A基因突变;原发性辅酶Q10缺乏症;共济失调;癫痫发作;常染色体隐形遗传病
原发性辅酶Q10缺乏症4型(primary coenzyme Q10 deficiency-4, COQ10D4)是由辅酶Q8A(coenzyme Q8A, COQ8A)基因突变导致的常染色体隐性遗传病,为可治性疾病。该疾病临床表现具有高度的异质性,主要表现为进行性步态共济失调、运动障碍,甚至可伴有认知功能障碍及癫痫等。本文报告1例由COQ8A新的基因位点突变所致原发性辅酶Q10缺乏症。本研究根据《赫尔辛基宣言》进行,并得到成都医学院第一附属医院伦理委员会的批准(编号:LXKY00130)。
1 临床资料
患者,男,26岁,汉,未婚。因“间断四肢抽搐伴意识障碍3 h”收入成都医学院第一附属医院治疗。患者清晨睡眠中被发现双上肢强直、双下肢抽搐,双眼上翻,无牙关紧闭、大小便失禁及偏侧肢体瘫痪,持续5 min左右抽搐症状缓解,10 min左右意识逐渐恢复,数分钟后再次出现抽搐,症状同前,共发生3次。120院前处置予以静脉推注“地西泮注射液”对症治疗后,抽搐未再发作,遗留意识模糊。既往史:10岁左右开始出现运动不耐受,对比同龄人运动明显减少,言语减慢,稍含糊,伴停顿,近5年逐渐间断出现头部不自主震颤及双手意向性震颤,紧张时明显,未诊治。家族史:父母体健(非近亲结婚),另外一个孩子(其兄长)半岁时夭折(具体不详)。个人史无特殊。入院查体:生命体征平稳,神志清楚,语速较慢、偶有结巴,余脑神经查体未见异常,四肢肌力5级,肌张力适中,四肢腱反射正常,双侧指鼻试验欠稳准,跟膝胫试验尚可,偶有双上肢活动性震颤,偶有头部及肢体震颤,紧张时加重,双侧病理征阴性,颈软,克氏征阴性。辅助检查:共济失调评估和评级量表(scale for the assessment and rating of ataxia,SARA)评分4分(步态1分,站姿1分,指鼻试验1分,构音不良1分),蒙特利尔认知评估量表28分,简易智能精神状态检查量表28分,焦虑抑郁自评量表正常。颅脑MRI可见小脑萎缩,见图1。视频脑电图监测未见明显异常。胸部CT、心脏彩超、心电图未见异常。肌红蛋白420.90 μg/L(参考值:<110 μg/L),肌酸激酶724 U/L(参考值:≤200 U/L),血清乳酸、血常规、肝肾功能、甲状腺功能、铜蓝蛋白、维生素B12、输血全套均正常,脑脊液检查正常。考虑患者少年起病、存在癫痫发作及隐匿进展的共济失调症状,遂完善全外显子基因测序,结果示COQ8A基因存在复合杂合突变,分别为NM_020247.5:exon8:c.1009G>A(p.A337T)(来自父亲)和NM_020247.5:exon8:c.1078delC(p.Q360Sfs*20)(来自母亲),根据美国医学遗传学与基因组学学会(the American College of Medical Genetics and Genomics, ACMG)评级,前者突变为PM2_P+PP3_M(临床意义未明),后者为PVS1+PM2_P(可能致病性)。家系图见图2,基因测序峰图分别见图3。
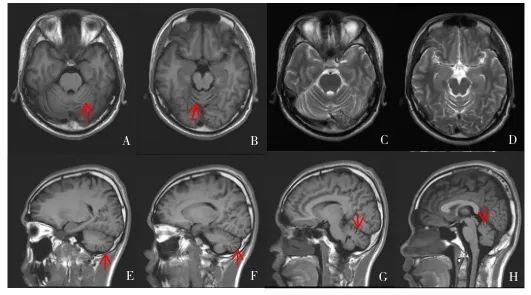
图1 患者颅脑MRI检查 小脑轻度萎缩,红色箭头所示。A~D为横断位图,E~H为矢状位图,A、B为T1加权成像,C、D为T2加权成像。Fig.1 Brain MRI examination of the patient’s brain
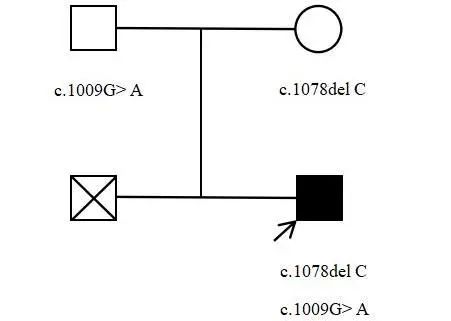
图2 患者家系图 ■为先证患者。□为男性,○为女性,x为已死亡。Fig.2 Pedigree chart of the patient
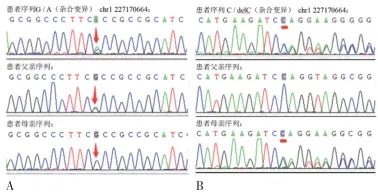
图3 测序峰图 A. c.1009G>A(p.A337T);B. c. 1078delC(p.Q360Sfs*20)。Fig.3 Sequencing peak map
考虑诊断原发性辅酶Q10缺乏症。停用抗癫痫药物,补充辅酶Q10治疗,100 mg/次、3次/d。1年后随访,患者头部震颤较前减轻,未再出现癫痫发作,运动耐量改善不明显,SARA评分3分(步态1分,站姿0分,指鼻试验1分,构音不良1分)。
2 讨论
辅酶Q10是人类线粒体氧化磷酸化系统中的重要辅因子,其生物合成是多种酶促反应参与的复杂过程,如果其合成酶的编码基因发生缺陷,可导致辅酶Q10合成下降,进而影响线粒体功能,引起一系列临床表现。若突变基因直接参与辅酶Q10生物合成,为原发性辅酶Q10缺乏症。原发性辅酶Q10缺乏症自1989年首次报告至今,已发现11个突变基因,分别为PDSS1、PDSS2、COQ2、COQ4、COQ5、COQ6、COQ7、COQ8A(ADCK3/CABC1)、COQ8B(ADCK4)、COQ9和HPDL[1-3]。
COQ10D4也称为COQ8A共济失调、常染色体隐性遗传性小脑性共济失调2型(autosomal recessive cerebellar ataxia type 2,ARCA2)和脊髓小脑性共济失调-9(spinocerebellar ataxia-9,SCAR9),于2004年首次报告[4]。其流行情况尚不清楚,目前全球报道有100余例。发病年龄从婴儿到中年不等[5],50%的患者在6岁之前发病,所有患者在45岁之前出现症状[6]。本文报告的患者10岁左右发病。
COQ10D4临床表现具有高度的异质性,主要累及小脑和骨骼肌,而肾脏相对不受影响[7]。常见的临床表现是进行性步态共济失调[8]和运动障碍[6]。这可能与COQ8A在浦肯野细胞中高表达相关[9]。部分患者可出现癫痫发作、类中风发作、认知功能障碍、眼球受累、视力下降、感音神经性听力损失、抑郁症和弓形足[6]。早发型儿童患者通常伴有认知功能障碍[10]。而青少年患者常出现癫痫和运动不耐受[11]。本例患者认知功能相对正常,具有小脑性共济失调、运动不耐受及癫痫发作的临床症状,症状较典型。
肌肉活检中测定辅酶Q10水平是辅酶Q10缺乏症的金标准[5],常规实验室检查可能检测到乳酸和肌酸激酶水平升高,辅酶Q10下降。部分患者可伴有低磷血症[12]。同时影像学检查中,小脑萎缩几乎是一个普遍的现象,2020年TRASCHÜTZ等[6]发现约94%(51/54)的患者存在小脑萎缩,但疾病早期可能表现不明显。疾病严重时可伴有全脑萎缩[8],部分患者可伴有胸腰椎侧弯[5]。本例患者颅脑影像存在轻度小脑萎缩,肌红蛋白及肌酸激酶升高,不排除癫痫发作时肌肉损伤导致。遗憾的是,本例患者未进行辅酶Q10水平检测,且拒绝肌肉活检。通过检测COQ8A基因来确定致病变异来源,本例患者检查测出NM_020247.5:exon8:c.1009G>A(p.A337T)和NM_020247.5:exon8:c.1078delC(p.Q360Sfs*20),均为首次发现,本病例补充了COQ8A基因变异位点。
对COQ10D4较为公认的治疗方案主要是补充辅酶Q10。补充后可以恢复浦肯野细胞形态、线粒体功能障碍和钙稳态[9]。然而,不同患者的治疗效果差别很大。两项研究显示分别有11例(50%)和13例(43%)患者补充辅酶Q10后症状有好转[6,13]。辅酶Q10的剂量范围为5~3000 mg/(kg·d)[5]。同多数线粒体疾病一样,COQ10D4患者需避免使用丙戊酸钠[11]。本例患者停用丙戊酸钠后未再出现癫痫发作,且补充辅酶Q10后,SARA评分下降1分,证明补充辅酶Q10有效,但仍需长时间随访观察。
本文首次报道COQ8A基因一种新的杂合变异所导致的原发性辅酶Q10缺乏症,该患者具有显著的临床特征,包括共济失调、头部震颤、运动不耐受和癫痫发作。
参考文献:
1. PRASUHN J, GÖTTLICH M, EBELING B, et al. The cerebellar bioenergetic state predicts treatment response in COQ8A-related ataxia[J]. Parkinsonism Relat Disord, 2022, 99: 91-95.
2. BANH R S, KIM E S, SPILLIER Q, et al. The polar oxy-metabolome reveals the 4-hydroxymandelate CoQ10 synthesis pathway[J]. Nature, 2021, 597: 420-425.
3. MÜNCH J, PRASUHN J, LAUGWITZ L, et al. Neuroimaging in primary coenzyme-Q-deficiency Disorders[J]. Antioxidants (Basel), 2023, 12(3): 718.
4. AURÉ K, BENOIST J F, OGIER D B H, et al. Progression despite replacement of a myopathic form of coenzyme Q10 defect[J]. Neurology, 2004, 63: 727-729.
5. ZHANG L W, ASHIZAWA T, PENG D T. Primary coenzyme Q10 deficiency due to COQ8A gene mutations[J]. Mol Genet Genomic Med, 2020, 8: e1420.
6. TRASCHÜTZ A, SCHIRINZI T, LAUGWITZ L, et al. Clinico-genetic, imaging and molecular delineation of COQ8A-ataxia: A multicenter study of 59 patients[J]. Ann Neurol, 2020, 88: 251-263.
7. STEFELY J A, LICITRA F, LAREDJ L, et al. Cerebellar ataxia and coenzyme Q deficiency through loss of unorthodox kinase activity[J]. Mol Cell, 2016, 63: 608-620.
8. MIGNOT C, APARTIS E, DURR A, et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression[J]. Orphanet J Rare Dis, 2013, 8: 173.
9. MANOLARAS I, DEL B A, GRISO O, et al. Mitochondrial dysfunction and calcium dysregulation in COQ8A-ataxia Purkinje neurons are rescued by CoQ10 treatment[J]. Brain, 2023, 146: 3836-3850.
10. BLUMKIN L, LESHINSKY-SILVER E, ZEREM A, et al. Heterozygous mutations in the ADCK3 gene in siblings with cerebellar atrophy and extreme phenotypic variability[J]. JIMD Rep, 2014, 12: 103-107.
11. ASHRAFI M R, HAGHIGHI R, BADY R S, et al. Epilepsia partialis continua a clinical feature of a missense variant in the ADCK3 gene and poor response to therapy[J]. J Mol Neurosci, 2022, 72: 1125-1132.
12. HAJI S, MIYAMOTO R, MORINO H, et al. Autosomal recessive spinocerebellar ataxia type 9 with a response to phosphate repletion: A case report[J]. Neurol Genet, 2023, 9(3): e200070.
13. CHANG A, RUIZ-LOPEZ M, SLOW E, et al. ADCK3-related coenzyme Q10 deficiency: A potentially treatable genetic disease[J]. Mov Disord Clin Pract, 2018, 5: 635-639.
【引用格式】朱姝,余凯,邹显巍. COQ8A基因突变所致原发性辅酶Q10缺乏症1例[J]. 中国神经精神疾病杂志,2024,50(10):605-607.
【Cite this article】ZHU S ,YU K,ZOU X W.Case report of primary coenzyme Q10 deficiency caused by COQ8A gene mutation[J]. Chin J Nervous Mental Dis,2024,50(10):605-607.
DOI:10.3969/j.issn.1002-0152.2024.010.007

