Fahr病(FD):原发性家族性脑钙化
时间:2025-01-16 12:11:01 热度:37.1℃ 作者:网络
论坛导读:Fahr病(Fahr’s disease,FD),也称为家族性特发性基底神经节钙化或双侧苍白球齿状突钙化症,是一种罕见的以丘脑、基底神经节、大脑皮质和小脑齿状核内异常血管钙沉积为特征的疾病。卡尔·西奥多·法尔( Karl Theodor Fahr)于1930年首次报道了这一现象,临床包括帕金森症在内的运动障碍是FD最常见的症状,尽管已知也存在惊厥和认知障碍。
Fahr病也称为原发性家族性脑钙化(Primary familial brain calcification,PFBC),文献中至少提过35种名称。该病的主要特征是双侧豆状核钙化,也可累及丘脑,尾状核和齿状核。小脑,脑干,半卵圆心和皮层下白质也可能受累。从流行病学来看,这是一种罕见的疾病,发病率低于百万分之一。然而,现有数据表明,目前估计的流行率为每1 000人中2.1至6.6人。Karl Theodor Fahr在1930年首次描述了FD。它主要影响30到50岁的人,但也可能在成年后发生。大多数病例表现为运动障碍或锥体外系症状,也有报道罕见以癫痫发作为首发症状。
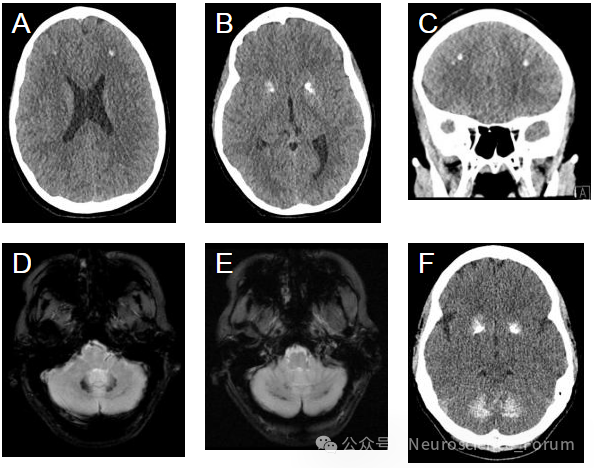
FD是一种涉及基底神经节和其他脑区的非动脉粥样硬化性脑内钙化的疾病。这种罕见的遗传性神经退行性疾病主要是作为常染色体显性性状遗传的,尽管偶发性或常染色体隐性发生也是可能的。FD的确切原因尚未完全阐明,在分子和遗传水平上的证据有限。虽然大多数FD病例与基因突变有关,但在大约三分之一的病例中,原因仍然不明。参与FD的关键初始基因包括8号染色体上SLC 20 a 2[钠依赖性磷酸盐转运蛋白2 (PiT-2)]和PDGFRB(血小板衍生生长因子受体β)基因的突变,这些基因对于维持血脑屏障的完整性非常重要。大多数研究表明SLC20A2基因突变(40%)导致钙和磷酸盐代谢失调,这通常在功能性消化不良的发病机制中起作用。此外,基因PDGFB(血小板衍生生长因子β)和XPR1(异嗜性和多变性逆转录病毒受体1)的突变也有关系。
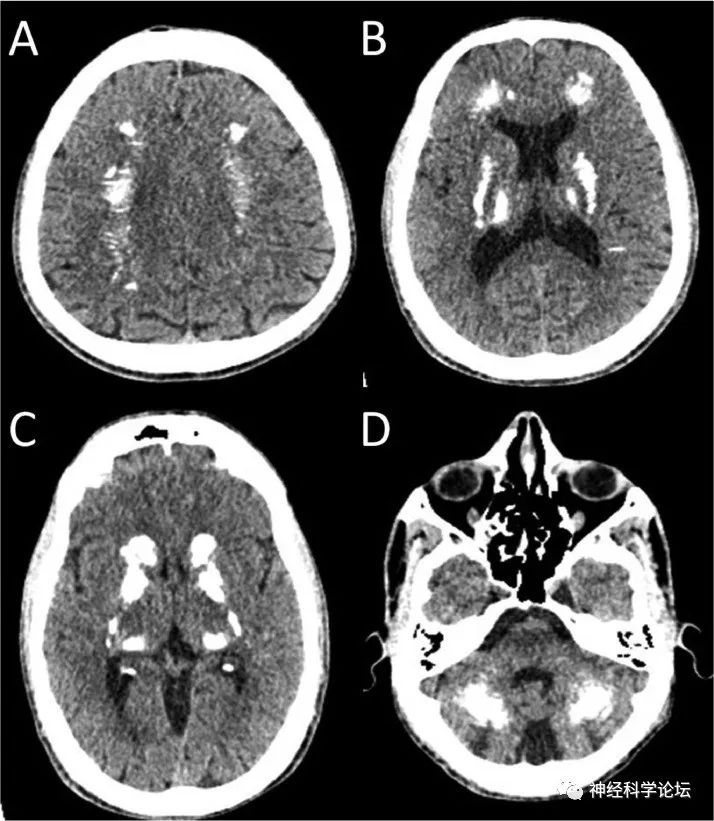
Dement Neurocogn Disord. 2023 Jul;22(3)
FD是一种罕见的遗传性或偶发性神经系统疾病,以大脑不同部位,特别是基底神经节和皮质下白质的双侧对称进行性钙化为特征。这是由于钙磷代谢的破坏和血脑屏障完整性的扰乱引起的异常钙沉积。考虑到病因,双侧条纹状多齿钙质沉着症可细分为四种形式,第一至第三种属于Fahr病,这意味着缺乏次要原因:1 .常染色体显性;2.家族性;3.散发的;4.次要的。常染色体显性形式涉及同一父母系中的不同人(先证者的父母或后代),似乎主要与染色体基因座14q48突变(IBCG1)相关。当同一个家庭的许多成员都受到影响时,就会诊断出家族形式,但这可以被认为是偶然的,而且没有特征认为它是一种遗传形式。当一个家庭只有一个人患病时,Fahr病是散发性的;当然,其他家庭成员缺乏症状不足以考虑散发病例,但充分的神经影像学研究应排除大脑钙化。
必须强调的是,随着年龄的增长,超过50岁,基底神经节一定程度的钙化可以被认为是“生理性的”。目前对Fahr病的研究已经证明这种病理没有特定的病因,可能的解释是继发于局部血屏障障碍或神经元钙代谢障碍的钙转移沉积,临床通常通过磷酸钙和碳酸钙形成的钙沉积来识别和诊断,这些钙化通常在基底神经节、丘脑、齿状核、海马、大脑皮质和皮质下白质中发现。基底神经节是最常受到影响的。这是一种罕见的遗传疾病,主要与染色体基因座14q48突变(IBCG1)相关,通常为显性遗传,但有零星病例报道。有报道了一例家族性基底神经节钙化的病例,这种钙化可能是由SLC20A2中的移码突变(c.1097delG p.G366fs *89)引起的。
FD修订诊断标准(Saleem et al,2013)
-
1.神经影像显示的基底神经节和其他脑区的双侧钙化。
-
2.进行性神经功能障碍,通常包括运动障碍和/或神经精神表现。
-
3.发病年龄通常在40或50岁,尽管这也可能出现在儿童期。
-
4.没有提示线粒体或代谢疾病或其他系统性疾病的生化异常和躯体特征
-
5.无传染性、毒性或创伤性原因。
-
6.家族史符合常染色体显性遗传。
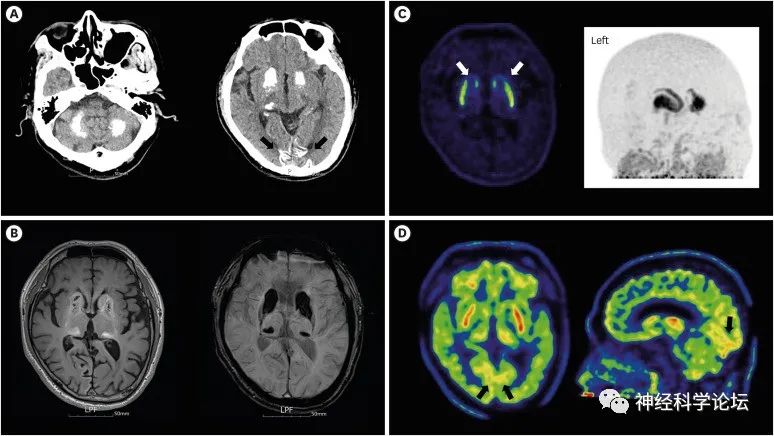
Radiol Case Rep. 2023 Mar 17;18(5)
FD以基底神经节和其他大脑和小脑结构的微血管的双侧进行性钙化为特征。FD患者中CT扫描中偶然发现基底神经节钙化的频率从80年代的0.24-0.75%上升到2000年的12.5 %。这种增加的原因一方面取决于CT扫描的广泛普及,另一方面取决于更大的新诊断标准(从孤立于苍白球的微小单侧钙化到大量双侧钙化)。
在鉴别诊断中还应考虑其他罕见的基底神经节金属沉积的遗传综合征,如铁和锰,因为它们在脑MRI上具有相似的特征,即使发病年龄通常早于PFBC,并且CT扫描可以帮助区分沉积的性质。其中,伴有脑铁积聚综合征(NBIA)、PKAN(泛酸激酶相关的神经变性,由于双等位基因PANK2突变)的神经变性可以表现为基底神经节内点状钙沉积超过潜在的铁积聚。在具有肌张力障碍和神经精神特征的患者的BPAN(β-推进器相关神经变性,由于WDR45突变)中也报告了一些钙化病例。其他成人发病的神经退行性疾病。此外,以颅内钙化为特征的罕见神经退行性疾病包括SCA20(小脑齿状核内孤立的钙沉积,无基底神经节参与)、NFT突变导致的神经铁蛋白病、多囊脂膜性骨发育不良伴硬化性白质脑病(Nasu哈科拉病)和弥漫性神经原纤维缠结伴钙化(Kosaka-Shibayama病)。
全外显子组测序(WES)和定制的NGS板是有用的诊断工具,用于识别与FD或其他以颅内钙化为特征的罕见疾病相关的基因突变,从而允许对受影响的受试者进行精确的分子诊断和遗传咨询。即使在无症状受试者中也具有该疾病的标志,即基底神经节钙化的放射学证据,可以帮助确定通过遗传分析突出的未知重要性(VUS)变异的致病性。为此,在患者亲属中进行基因检测时,应始终伴有脑部CT扫描。临床医生必须考虑中年患者急性精神状态改变的精神病学和医学原因。如果患者出现多次发作和症状恶化,每次急性变化发作都应保证进行全面的实验室检查和神经影像学检查,以排除可能的病因。在FD开发针对改变的通路的疾病特异性药物是该领域的一个主要挑战,该领域与其他几种以人体组织中钙异常沉积为特征的遗传性人类疾病部分重叠。目前已发现7个致病基因,其中4个为显性基因(SLC20A2,PDGFB,PDGFRB,XPR1),3个为隐性基因(MYORG,JAM2,CMPK2)。临床表现范围从无症状受试者到运动障碍、认知能力下降和精神障碍(单独或多种组合)。在所有已知的遗传形式中,钙沉积的放射学模式是相似的,但脑桥中央钙化和小脑萎缩高度提示MYORG突变,广泛的皮质钙化与JAM2突变相关。
综上所述,FD目前被认为是由于神经血管单元(NVU)的功能改变,其中异常的钙磷代谢、周细胞的功能和显微解剖学改变以及线粒体改变导致血脑屏障(BBB)功能障碍,并产生成骨环境,周围星形胶质细胞活化和进行性神经变性。目前,没有缓解疾病的药物或钙螯合剂可用,只能提供对症治疗。
参考文献
Savino E, Soavi C, Capatti E, Borrelli M, Vigna GB, Passaro A, Zuliani G. Bilateral strio-pallido-dentate calcinosis (Fahr's disease): report of seven cases and revision of literature. BMC Neurol. 2016 Sep 8;16(1):165. doi: 10.1186/s12883-016-0693-1.
Carecchio M, Mainardi M, Bonato G. The clinical and genetic spectrum of primary familial brain calcification. J Neurol. 2023 Jun;270(6):3270-3277. doi: 10.1007/s00415-023-11650-0.
Dennis AC, Nwabueze C, Banu F, Nisenoff CD, Olupona T. Bilateral Basal Ganglia Calcifications Manifesting as Psychosis With Manic Features: A Case Report on Fahr's Syndrome. Cureus. 2023 Feb 2;15(2):e34547. doi: 10.7759/cureus.34547.
Li M, Fu Q, Xiang L, Zheng Y, Ping W, Cao Y. SLC20A2-Associated Idiopathic basal ganglia calcification (Fahr disease): a case family report. BMC Neurol. 2022 Nov 17;22(1):438. doi: 10.1186/s12883-022-02973-y.

