【JAMA】综述:真性红细胞增多症的诊断和治疗
时间:2024-11-27 12:00:36 热度:37.1℃ 作者:网络
真性红细胞增多症
真性红细胞增多症(PV)是一种慢性骨髓增殖性肿瘤,其特征为髓系细胞的克隆性增殖导致红细胞容量增加。超过95%的PV患者存在获得性活化JAK2基因变异。在美国,PV的年发病率估计约为0.5-4.0/10万人,估计患病人数为65000人。诊断时的中位年龄为65岁,男性的发病率略高(男性与女性的发病率比为1.3-1.6)。PV的危险因素包括吸烟、暴露于电离辐射和某些环境危险因素(如苯),但大多数患者没有已知的毒性暴露。尽管JAK2基因变异是获得性的,但也可能存在遗传性易感因素促进PV的发展,例如一级亲属中患病的风险约为5倍。
PV中导致发病和死亡的主要并发症为动脉和静脉血栓,发生率分别有16%和7%,可在诊断前或诊断时存在。诊断PV后,动脉血栓的发生率还会增加12%,静脉血栓的发生率则增加9%。此外PV患者出血和瘀伤的风险增加,尤其是重度血小板增多(血小板计数≥1000x109/L)和获得性血管性血友病患者。约 12.7%的PV患者会进展为骨髓纤维化,6.7%发展为急性髓系白血病(AML)。
JAMA近日发表综述,总结了有关PV诊断和治疗的现有证据,现整理供参考。

关于真性红细胞增多症(PV)的常见问题
如何区分PV和继发性红细胞增多症?
超过95%的PV患者存在JAK2基因序列变异,可用于区分PV与继发性红细胞增多症。此外,PV患者通常血清促红细胞生成素水平较低(或正常),而继发性红细胞增多症促红细胞生成素水平升高。
PV的潜在并发症有哪些?
PV患者中约有30%会发生血栓,包括动脉血栓和静脉血栓,与发病率和死亡率的增加相关。PV患者出血以及发生骨髓纤维化和AML的风险也增加。水源性瘙痒、红斑性肢痛和疲劳等症状也可能出现,对患者的生活质量产生负面影响。
如何治疗PV?
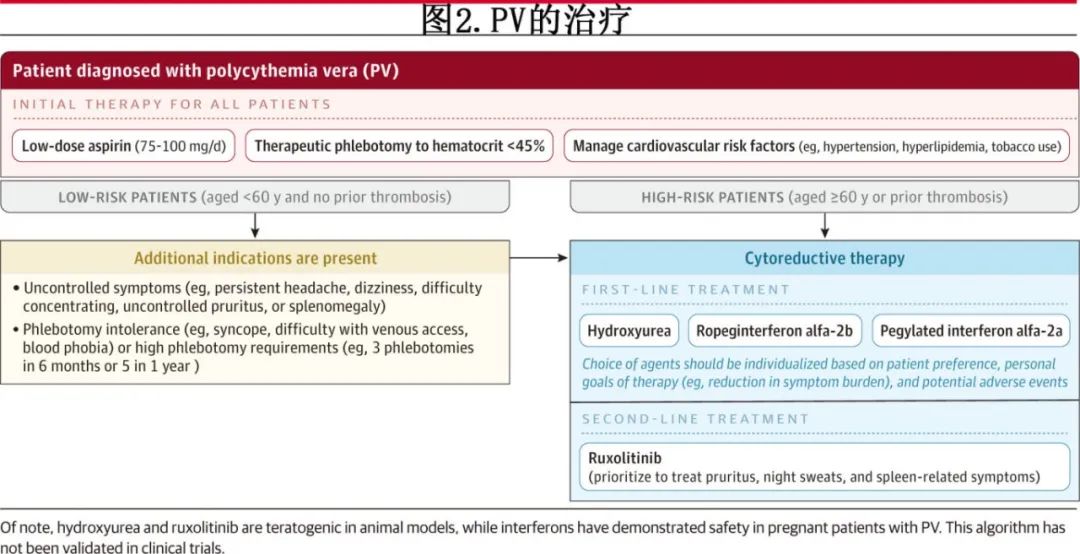
所有PV患者都应接受治疗性静脉放血,将红细胞压积降低至45%以下,并接受小剂量阿司匹林(每日75-100mg)治疗,除非有禁忌证。对于血栓风险高的患者或持续存在困扰症状的患者,可采用羟基脲或基于干扰素的疗法进行降细胞治疗。对于无法耐受或抵抗羟基脲的患者,JAK1/JAK2 抑制剂芦可替尼可作为二线治疗。
病理生理学
PV引起红细胞增多症(男性血红蛋白>16.5mg/dL,女性>16.0mg/dL),并伴有血清促红细胞生成素降低。骨髓增殖性表型的潜在原因为JAK2基因变异导致鸟嘌呤被胸腺嘧啶取代,导致假激酶结构域14 (JAK2V617F)内密码子617处缬氨酸被苯丙氨酸取代,JAK2 基因变异会激活包括 JAK-STAT在内的信号级联反应,并增加红系祖细胞对促红细胞生成素的敏感性。由于在多能性造血干细胞中检测到了 JAK2 V617F 序列变异,它也可能导致白细胞增多和血小板增多。其他影响外显子 12 的 JAK2 功能获得性序列变异可以在缺乏 JAK2 V617F 的真性红细胞增多症患者中检测到,特别是在没有血小板增多症或白细胞增多症的患者中。
JAK2 序列变异可能在生命早期出现,但基因谱系研究表明出现 JAK2 序列变异到被诊断PV之间的平均时间为 30 年(11-54 年)。在丹麦无血细胞计数异常的一般人群中,约 3.1%检测到 JAK2 V617F 变异。对于这些人,JAK2 序列变异可能代表潜能未定的克隆性造血,与无序列变异者相比,其患冠心病的风险增加(风险比[HR]为 12.0)。
PV患者发生血栓事件的风险增加是多因素的,包括由于红细胞数量增加导致血液黏度增加。出血常与获得性血管性血友病相关,其可发生于血小板极度增多(血小板计数1000×109/L,见于 4%的PV患者)的情况下,服用阿司匹林或抗凝药可能会使其恶化。
临床表现
PV的症状包括对任何温度的水产生强烈瘙痒或刺痛感(水源性瘙痒),约三分之一的患者在确诊时可出现。其他临床发现包括红斑性肢痛(5.3%)、短暂性视力变化(14%)和脾肿大(36%),后者可能导致腹部不适。在确诊PV前,约 16%的患者存在动脉血栓,7%的患者存在静脉血栓,且可能发生在不寻常的部位,如内脏静脉。动脉血栓包括急性冠脉综合征(8.3%)和脑血管事件(7.7%),而静脉血栓包括深静脉血栓(4.2%)和肺栓塞(2.0%)。PV患者的静脉血栓可发生在非典型部位,包括内脏血管(所有血栓的 14.5%)、肝静脉(Budd-Chiari合征)或门静脉,以及脑静脉窦(2.7%)。
血栓的诊断与风险分层
WHO和ICC已建立了PV诊断标准,最新版是2022年版。红细胞增多是必需的诊断标准(其他骨髓增殖性疾病中不存在),男性血红蛋白>16.5g/dL和红细胞压积>49%,女性血红蛋白>16.0g/dL和红细胞压积>48%。
PV应与假性红细胞增多症区分开来,后者通常为非持续性,是由血浆容量收缩引起但红细胞容量正常(通过放射性标记法测量)。PV也应与继发性红细胞增多症区分开来,后者是由导致促红细胞生成素升高的疾病所引起,例如紫绀性心脏病、吸烟、阻塞性睡眠呼吸暂停、高海拔居住,或某些良性病症(如子宫肌瘤)或恶性肿瘤(如肾细胞癌或血管母细胞瘤)的分泌。用于提高运动表现的外源性雄激素和促红细胞生成素也与红细胞增多症有关。假性和继发性红细胞增多症不存在浆细胞增多症、白细胞增多症或血小板增多症。
许多红细胞增多症患者并非PV患者。在荷兰一项针对 ≥18 岁普通人群的队列研究(147168人)中,3.4%(男性中7.6%,女性中0.4%)患有红细胞增多症,其定义为男性血红蛋白大于16.5g/L,女性大于16.0g/L。在患有最严重红细胞增多症(男性>18.5 g/L,女性>16.5g/L)且伴有白细胞增多症(白细胞[WBC]≥11×109/L)和/或血小板增多症(血小板计数≥450×109/L)的子集与孤立性红细胞增多症(N=133人)按1:2匹配。在可评估JAK2 变异的患者中,患有重度红细胞增多症伴白细胞增多和/或血小板增多症的45 例中有 7例(15.6%)存在JAK2基因变异,而 88 例孤立性重度红细胞增多症患者中为0。
对于红细胞增多且红细胞生成素水平低的患者,以及所有在内脏血管或脑窦有血栓形的患者,都应接受 JAK2 基因变异检测。即使 JAK2 检测结果为阴性,但有约 2%的红细胞增多症患者是根据红细胞增多、红细胞生成素水平低以及不存在红细胞增多的继发性或遗传性病因(如红细胞生成素受体或von Hippel–Lindau基因序列变异)被重新诊断为PV。骨髓活检是WHO诊断PV的主要标准,但对于持续型绝对红细胞增多(通常为3个月或更长时间)且阈值高于诊断标准(男性血红蛋白浓度为18.5 g/dL或女性血红蛋白浓度为16.5 g/dL)和JAK2基因变异的患者,建议进行骨髓活检,但并非必需。图1概述了PV的诊断评估,中国PV诊疗指南与之一致。


患者在确诊PV后应接受血栓风险评估,可影响预后和治疗决策,包括关于降细胞治疗的决策。NCCN和ELN定义的高风险PV为年龄≥60 岁或有既往动脉或静脉血栓史的患者,低风险患者为年龄<60岁且无动脉或静脉血栓史的患者。基于一项针对 1638 例PV患者的研究的多因素模型,心血管事件相对风险(RR)因年龄>65 岁(年龄 66-75 岁,RR=2.08)及既往血栓史(RR=2.09)而增加。
PV患者的血小板增多程度并非血栓形成的风险因素。然而一项纳入 2510例PV 患者的前瞻性研究报告称,血栓事件与白细胞计数大于 11×109/L(HR=2.35,P<0.001)及红细胞压积大于 45%(HR=1.84,P=0.01)相关。在一项纳入533例接受羟基脲治疗的 PV 患者的研究中,每年需要≥3 次静脉放血的患者与每年需要 0 至 2 次静脉放血的患者相比,血栓的发生率更高(3 年时分别为 20.5%和5.3%;P<0.001)。
JAK2 V617F 等位基因负荷对PV患者的血栓风险和骨髓纤维化具有预后信息。该等位基因负荷是通过从细胞中提取 DNA 然后分析 JAK2 区域来量化包含 JAK2 V617F 基因变异的 JAK2 测序读段百分比(0%至 100%)获得。通常从外周血进行测量,但也可以从骨髓穿刺物评估。在一项856 例 PV 患者的观察性队列研究中,JAK2 V617F 等位基因负荷为 ≥50%与静脉血栓的风险增加有关(14.5% vs 2.4%;HR=3.8)。在一项 338 例 PV 患者的前瞻性研究中,JAK2 V617F 等位基因负荷≥50%也是进展为骨髓纤维化的风险因素(10.5% vs 1.2%;P=0.03)。
治疗
治疗性静脉放血
所有PV患者都应按照NCCN的建议(基于CYTO-PV研究),接受治疗性静脉放血,以维持红细胞压积低于 45%。CYTO-PV研究将365例 PV 患者随机分配至红细胞压积目标低于45%或45%-50%,通过治疗性静脉放血(68%的患者;开始时每隔一天或每周两次 250-500mL 血液,直至达到红细胞压积目标)和/或降细胞治疗(52.6%的患者;通常为羟基脲,初始剂量为每日500-1000mg)。红细胞压积小于45%组的主要终点(心血管死亡或严重血栓)为2.7%,而45%~50%组为9.8%(P=0.007)。尽管女性PV的基线血细胞压积水平通常低于男性,但针对女性PV患者的较低血细胞压积目标尚未得到严格评估。此外,尽管在高海拔地区(>1524 米)居住的PV患者中血栓可能更常见(慢性低氧会导致血细胞压积水平升高),但对于居住在高海拔地区的PV患者的治疗目标调整尚未进行前瞻性研究。
抗血小板药物
对于所有无抗血小板药物禁忌的PV患者,建议低剂量阿司匹林(每日75-100mg)。一项研究纳入518 例无其他抗血小板治疗指征(如中风预防或冠脉疾病)的 PV 患者,与安慰剂相比,随机分配至每日100mg阿司匹林组的患者在非致命性心肌梗死、非致命性中风、静脉血栓栓塞或心血管原因导致的死亡这一联合主要终点的风险降低(3.2% vs 7.9%;P=0.03)。但该研究并未报告总体或心血管特异性死亡率方面的显著差异,且阿司匹林组主要出血(1.2% vs 0.8%)的增加无统计学意义。
目前尚无关于使用氯吡格雷或其他 P2Y12 抑制剂进行初级血栓预防的已发表研究。对于有血栓事件的患者,目前尚无数据指导抗凝药的选择或持续时间。在PV患者中同时进行阿司匹林和抗凝治疗与出血风险增加相关(HR=5.83,P<0.001)。因此临床医生在处方抗凝药时往往会停止阿司匹林治疗,但该策略尚未进行前瞻性分析。
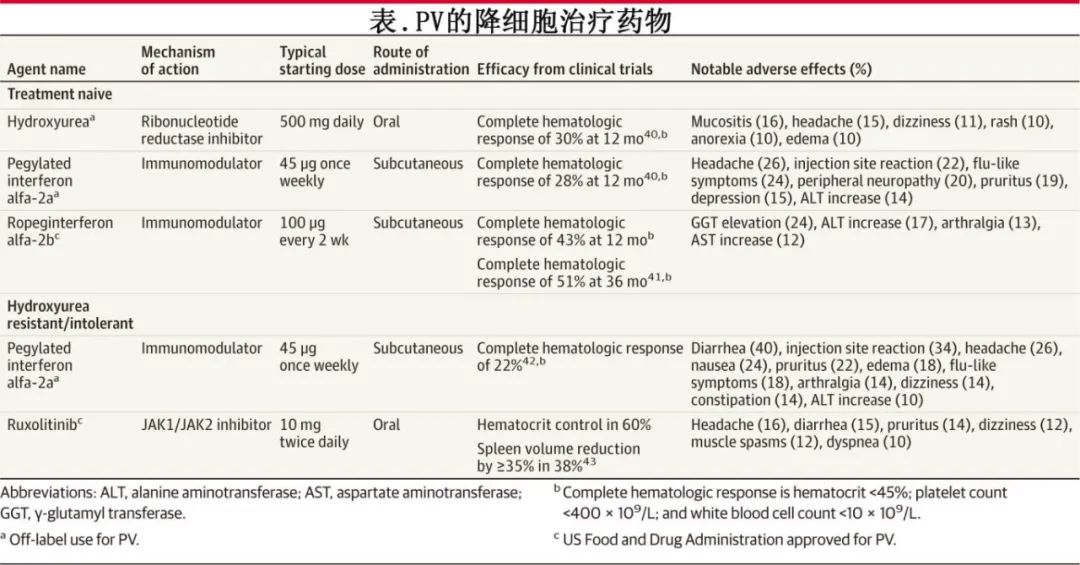
降细胞治疗
降细胞治疗包括羟基脲、干扰素制剂(罗格干扰素α-2b 和聚乙二醇干扰素α-2a)以及芦可替尼(表),可降低血细胞计数并减少血栓风险。降细胞治疗可在阿司匹林和治疗性静脉放血的基础上使用,适用于血栓风险高的患者(例如>60 岁或既往有动脉或静脉血栓史的患者),对于低风险PV患者,如果出现持续头痛、头晕、难以集中注意力、瘙痒未控制或使用抗血小板治疗和治疗性静脉放血的情况下仍有脾肿大,也可考虑使用。在一项纳入 127例低风险PV患者的随机研究中,治疗24 个月后,静脉放血和阿司匹林组中-重度症状的发生率从 43%增加到 67%,而随机分配至罗格干扰素α-2b、静脉放血和阿司匹林组的患者中-重度症状从 39%减少到 33%。
NCCN和ELN指南也建议,对持续存在白细胞增多症和/或血小板增多症的低风险PV患者进行降细胞治疗,但尚未完全确定启动治疗的白细胞和血小板阈值,以及降细胞治疗对白细胞增多症或血小板增多症患者预防血栓的疗效。接受降细胞治疗的患者应继续服用阿司匹林,并进行治疗性静脉放血,目标为红细胞压积低于 45%。
NCCN 指南建议高风险PV患者使用羟基脲或干扰素制剂。药物的选择应根据患者的偏好、治疗的个人目标(例如减轻症状负担),及药物相关的潜在不良事件进行个体化选择。
羟基脲
羟基脲是一种每日口服药物,通常起始剂量为每日两次,每次 500mg,可逐渐增加至每日两次,每次 1000 mg。羟基脲通常耐受性良好,但由于在动物研究中具有致畸作用,因此禁用于孕妇或备孕女性。羟基脲的不良反应可能包括黏膜炎、皮肤溃疡和头发稀疏。此外基于一项127 例病例和 244 例对照的嵌套病例对照研究,羟基脲与非黑色素瘤皮肤癌的风险增加相关(比值比为 2.28)。
一项倾向评分匹配回顾性分析报告称,在接受羟基脲治疗的 681 例PV患者中,与接受静脉放血治疗的 342 例 PV 患者相比,接受羟基脲治疗的患者发生致命和非致命心血管事件的总数显著降低(每 100 人年 3.0 例vs 5.8 例;P=0.002)。然而该获益仅见于血栓风险高的 PV 患者。
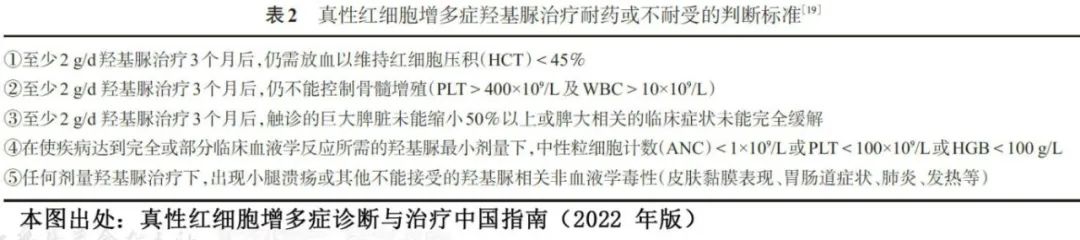
约 15%的PV患者对羟基脲不耐受或耐药,表现为需要治疗性静脉放血;血小板计数大于 400×109/L;白细胞计数大于10×109/L;在至少 3 个月的羟基脲治疗(每日最低剂量为 2000mg)后未缓解脾肿大相关症状;或者在达到完全血液学反应(红细胞压积<45%;血小板计数<400×109/L;白细胞计数<10×109/L)所需的最低剂量下出现中性粒细胞减少症、贫血或血小板减少症。对羟基脲产生耐药性或不耐受的患者应接受二线治疗,如干扰素或芦可替尼治疗。
干扰素
美国有两种PV的干扰素制剂:罗普格特干扰素α-2b(美国FDA批准)和聚乙二醇干扰素α-2a(超说明书使用)。罗普格特干扰素α-2b 每两周皮下注射一次,聚乙二醇干扰素α-2a 每周皮下注射一次。干扰素的剂量需逐渐调整以达到完全血液学反应。对于怀孕或有怀孕计划的 PV 患者,聚乙二醇干扰素α-2a是安全的。
干扰素的使用与抑郁症和焦虑症的风险增加相关,因此应避免在患有这些心理健康疾病的患者中使用。干扰素也不应用于患有严重或未治疗的自身免疫性疾病,如类风湿性关节炎和系统性红斑狼疮,因其存在加重这些疾病的风险。干扰素的不良反应可能包括头痛、注射部位反应、血清丙氨酸氨基转移酶升高、周围神经病变以及流感样症状(见表)。
在一项纳入 127 例低风险PV患者的开放性标签研究中,随机接受罗格干扰素α-2b 联合放血的患者(81.3%)中,血细胞压积低于 45%且无疾病进展这一主要终点更为常见,而仅接受放血的患者中为 59.7%(P<0.001)。与单纯放血和阿司匹林相比,罗格干扰素α-2b 在 12 个月内也与疾病进展的减少有关(0% vs 12.7%;P<0.001),但对照组中的疾病进展事件主要为血小板增多症(9.5%),且两组中均无患者进展为骨髓纤维化。罗格干扰素α-2b 组不良事件更常见,包括 9%的患者出现 3-4 级中性粒细胞减少症。在一项纳入 50 例对羟基脲耐药或不耐受的PV患者的单臂干预试验中,聚乙二醇干扰素α-2a治疗后60%的患者在 12 个月时出现完全血液学反应。
比较羟基脲与基于干扰素的治疗的研究
两项随机研究对比了在PV患者中羟基脲与基于干扰素的治疗。骨髓增殖性肿瘤研究联盟(MPN-RC)112 研究将 87 例PV患者随机分配至聚乙二醇干扰素α-2a 组或羟基脲组,两组患者在 12 个月完全缓解(定义为完全血液学反应及脾肿大和疾病相关症状[如微血管症状、头痛和瘙痒]缓解)方面无显著差异(28% vs 30%[P=0.86])。聚乙二醇干扰素α-2a 组轻-中度抑郁症更为常见(15%),而羟基脲组为 3%。
一项纳入 254 例患有早期PV(定义为未使用过降细胞治疗或使用羟基脲治疗少于 3 年)患者的 3 期随机研究报告称,罗格干扰素α-2b 与羟基脲治疗 的12 个月时完全血液学反应率无差异(43% vs 46%;P=0.63)。然而在参与研究扩展阶段的 171 例患者中,罗格干扰素α-2b 相对于羟基脲的 36 个月血液学反应率有所改善(67/95[71%] vs 38/74[51%];P=0.01)。研究结束时对 169例患者进行随访,因其在接下来的 5 年中继续接受治疗;罗格干扰素α-2b 组无事件生存率(血栓形成、疾病转化或死亡)高达 94%,而对照组为 82%(P=0.04)。
芦可替尼
JAK1/JAK2 抑制剂芦可替尼是一种口服药物,通常起始剂量为每日两次,每次 10mg,可逐渐增加至每日两次,每次 25 mg。芦可替尼总体耐受性良好,但与体重增加和带状疱疹感染风险增加相关(约 4%的治疗患者),可能是由于芦可替尼的免疫抑制作用。因此建议在开始使用芦可替尼之前接种带状疱疹重组疫苗。芦可替尼还与非黑色素瘤皮肤癌风险增加相关(接受芦可替尼治疗的患者中5.1/100患者/年,而接受其他治疗的患者为 2.7)。
多项随机临床研究表明,芦可替尼对接受过羟基脲治疗的患者有益。一项纳入 222 例PV伴脾肿大患者的研究中,患者对羟基脲不耐受或抵抗,随机分配接受芦可替尼治疗或研究者选择的降细胞治疗。在接受治疗的患者中,芦可替尼组20.9%达到主要复合终点,即红细胞压积控制(<45%)和脾脏大小缩小(在 32 周时通过影像学检查至少减少 35%),而标准治疗组为0.9%(羟基脲、干扰素、来那度胺、沙利度胺、聚乙二醇干扰素或无治疗)。在 14 项骨髓增殖性肿瘤症状评估量表总症状评分中减少>50%方面,两组分别为49%和5%,且改善主要是在瘙痒、夜间出汗和早期饱腹感方面。此外,一项纳入 173 例无脾肿大的PV患者的开放性研究报告称,芦可替尼组与标准治疗组相比,第 28 周红细胞压积控制情况有显著改善(62% vs 19%;P<0.001)。
2023 年的一项研究将 180 例对羟基脲治疗无效或不耐受的PV患者随机分组,分别接受芦可替尼治疗和最佳可用治疗。结果显示,1 年时完全血液学反应和脾脏大小恢复正常的比例为43% vs 26%(P=0.02),症状改善率为61% vs 30%(P=0.001)。芦可替尼在无事件生存期(主要血栓形成、出血、转化和死亡)方面也具有优势(HR=0.58;P=0.03),主要是由于血栓事件数量减少(HR=0.56;P=0.05)。此外,芦可替尼治疗后突变反应率(mutational response,JAK2 V617F 等位基因负荷减少 50%以上)更高(56% vs 25%;P<0.001),其与随访期间进展率降低(39% vs 11%;P=0.001)及无事件生存期(54% vs 25%;P=0.001)和总体生存期(24% vs 8%;P=0.01)改善相关。
然而,一项纳入 110 例PV(羟基脲控制良好,但存在PV相关症状)的3b 期双盲研究中,在 16 周主要终点(患者症状评分减少 50%)方面,芦可替尼组与安慰剂组相比没有显著差异,主要终点包括疲劳、瘙痒、肌肉疼痛、夜间出汗和清醒时出汗(芦可替尼组 43.3%,安慰剂组 29.6%,P=0.14])。
图 2 详细介绍了PV的当前治疗方案。
预后
在一项国际多中心研究(7家中心1545例患者)中,诊断时的中位年龄为 61 岁,在达到中位生存期的 5 家中心中, PV诊断后的中位生存期为 14.1 至 27.6 年。1当将所有 7 家中心纳入生存分析时,中位生存期为 18.9 年,与来自美国总人口中相同年龄和性别的个体相比无差异(P=0.14)。与生存期缩短相关的因素包括年龄≥67岁(HR=8.5)、白细胞计数≥15×109/L(HR=2.2)以及既往静脉血栓史(HR=1.8)。在一项评估 1967 年至 2017 年患者的美国队列研究中,诊断时年龄小于 40 岁的患者(361例)中位总生存期为 37岁。此外在一项纳入 404例PV的研究中,控制前体 mRNA 剪接的富含丝氨酸/精氨酸的剪接因子 2(SRSF2)基因变异与生存期缩短相关(相对于没有这些序列变异的患者,HR=13.3)。
PV可能会发展为骨髓纤维化和AML。在一项纳入 267 例PV患者的队列研究中,随访11.8 年,12.7%的患者发展为骨髓纤维化,6.7%的患者发展为AML;转化为骨髓纤维化后的生存期约为 8.1 年。与骨髓纤维化或AML风险增加相关的因素包括持续白细胞增多、JAK2V617F 等位基因负荷≥50%(转化为骨髓纤维化或AML的风险为 10.5% vs 1%)、SRSF2 或 IDH2 的序列变异以及 1q 染色体获得。骨髓活检虽然并非诊断真性红细胞增多症所必需,但可能识别出基线骨髓中的纤维化(bone marrow fibrosis),其与进展为骨髓纤维化(myelofibrosis)的风险增加相关。
总结
PV是一种克隆性骨髓增殖性肿瘤,会导致红细胞增多,通常与 JAK2 基因变异相关。PV患者发生动脉和静脉血栓、出血、中风、心肌梗死、骨髓纤维化和AML的风险增加。所有PV患者都应服用阿司匹林(如无禁忌),并进行治疗性静脉放血,以将红细胞压积维持在45%以下,降低血栓风险。对于血栓形成风险高的PV患者,建议采用降细胞治疗,如羟基脲或干扰素。
参考文献
Tremblay D,et al.Diagnosis and Treatment of Polycythemia Vera.JAMA . 2024 Nov 18. doi: 10.1001/jama.2024.20377.

