昭衍·讲堂 基因治疗主题系列:从CDMO到临床检测,CGT疗法的希望、挑战和未来
时间:2022-11-18 20:51:44 热度:37.1℃ 作者:网络
从问世到叫停再到重回视野,基因治疗发展历经“乐观与热情—失望与怀疑—理性与挑战”。
1990年,患有重症联合免疫缺陷病(severe combined immunodeficiency diease,SCID)的4岁女孩阿莎提·德席尔瓦,被输入利用逆转录病毒将正确编码腺苷脱氨酶的ADA(adenosine deaminase)基因导入离体培养的患者淋巴细胞,该治疗有效恢复患者体内ADA的合成。此次尝试的成功,无疑造就了之后大量基因治疗临床试验的接踵而至。
然而,1999年,18岁的美国男生在基因治疗过程中接受腺病毒注射几天后,由于免疫系统对腺病毒过度反应导致器官衰竭而不幸死亡;2002年,接受了逆转录病毒注射的两名SCID患者继发白血病。这导致FDA于2003年叫停了所有利用逆转录病毒的临床试验。但在三个月审核后,FDA通过权衡认为基因治疗利大于弊,于是决定再次开启逆转录病毒临床试验。
此后,基因治疗的安全性研究便成了科学家们一直努力的方向,不过当前基因治疗领域面临的挑战绝不止于此。
技术、生产、商业化 基因治疗面临多重挑战
资料显示,广义的细胞和基因治疗(Cellular and Gene Therapy, CGT)包括细胞治疗和基因治疗。目前业内公认的CGT主要是指基因治疗,即通过基因添加、基因修正、基因沉默等方式修饰个体基因的表达或修复异常基因,达到治愈疾病目的的疗法。
其核心在于精准打击疾病根源——异常DNA,通过调控DNA来改变遗传信息传递,从而改变蛋白质性状,实现从根源上治疗疾病,是一种根本性的治疗策略。
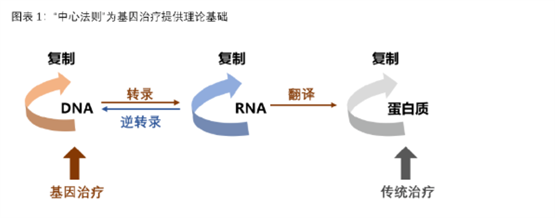
根据治疗途径,基因治疗可分为体内基因治疗和体外基因治疗。其中体内基因治疗是指将携带治疗性基因的病毒或非病毒载体直接递送到患者体内,体外基因治疗则指将患者的细胞在体外进行遗传修饰后回输。
无论是在体外还是体内进行基因改造,基因治疗的三大共性步骤为:
·核酸序列的设计与合成
·将目标序列递送至细胞中(体内或体外)
·工业化生产
其中,核酸序列的设计与合成难度较小分子靶向药和单抗药物更低。因此一旦研发出一个安全高效的递送系统,基因治疗产品的研发难度反而更低,研发成功率更高。
递送方式包括病毒载体和非病毒载体两大类,非病毒载体的工业化级别放大相对容易,但病毒载体的规模化生产仍面临一定的瓶颈。
据了解,病毒载体生产的上游(USP)主要包括病毒构建(转染)和细胞扩增培养,难点在于如何减少瞬时转染所需的质粒数量以及提高转染效率、如何提高细胞培养密度、扩大产能等。
其下游(DSP)则包括细胞裂解、澄清、浓缩、洗滤、层析纯化、精制及除菌过滤等多项工艺,主要难点在层析纯化环节。目前病毒载体DSP整体收率仅为20-30%,难点在于USP中存在的空壳病毒(不含有治疗基因但会引起免疫反应)。
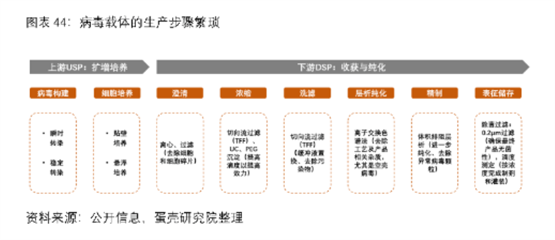
此外,基因治疗还存在如何提升转导效率、提高靶向组织特异性、AAV载体容量小、Cas9酶自身的大尺寸、易聚集或沉淀、脱靶效应(CRISPR编辑系统对体内非目标区域的DNA双链进行了错误切割,可能引发安全性问题)、以及可能面临的免疫障碍(适应性免疫反应和先天免疫反应)等多重挑战。
CGT领域多为初创公司 其研发生产高度依赖外包
基因治疗研发生产的高门槛间接使得专业化CRO/CDMO服务将成为基因治疗药物研发的刚性需求。
在研发方面,基因治疗药物具有高度复杂性。其与传统制药相比,基因治疗以基因功能研究为基础,目的为干预患者特定细胞内的蛋白表达或RNA表达;并且除肿瘤适应症外,大量药物研发管线系针对血友病、地中海贫血、部分眼科疾病等罕见遗传病,其药理、药效安全评价体系较新,行业成熟经验远少于传统药物。
并且目前基因治疗上市药物较少,药物研发经验不足,因此需要对基因功能进行深入研究,这就需要基因治疗CRO提供专业化的先导研究服务。
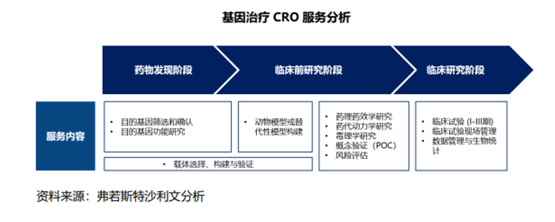
同样的,在生产方面,基因治疗药物生产涉及更多对象、纯化及测试分析,导致其生产周期更长、步骤更多、成本更高,决定了CDMO依旧也是不可或缺的外包模式。
具体来看,一方面基因治疗药物多处于早期开发阶段。由于基因治疗生产过程复杂、涉及多个环节的工艺开发和质控方法开发、容错率低、对于过程控制的要求非常严苛、整体难度较大,因而早期开发阶段药企更倾向于选择CDMO进行合作。
另一方面,全球病毒载体产能不足,CDMO供不应求。Oxford Biomedica公布的全球病毒载体市场规模预测数据显示,2020-2026年全球病毒载体市场规模将由12亿美元增长到28亿美元,2020-2026年CAGR为15.17%。弗若斯特沙利文分析数据显示,2025年全球基因治疗市场规模就将超过300亿,而正在开发的基因疗法中88%利用病毒载体进行传递,病毒载体的供应量是远远不够的。
值得注意的是,根据Roots Analysis数据,大约91%的基因治疗研发管线由中小企业贡献。由此可见,基因治疗领域初创公司占比较大,由于受到工艺开发能力、GMP生产经验、临床申报相关法规知识的限制,这些初创企业更多依赖专业的CRO和CDMO服务。
完成全球首个上市基因治疗项目系统评价 昭衍新药CGT疗法订单快速增长
作为中国最早从事药物非临床评价的民营CRO企业,北京昭衍新药研究中心股份有限公司(“昭衍新药”)具有符合国际规范的质量管理体系(CNAS/ILAC-MRA认证),具备中国NMPA、美国FDA、经合组织OECD、韩国MFDS、日本PMDA的GLP资质以及国际AAALAC和OLAW(动物福利)认证资质,评价资料满足全球药品注册要求。可以向客户提供CDMO、药理药效学研究、生物分析(DMPK)、药物安全性评价研究、临床检测等一站式服务。
在CGT疗法方面,公司针对CGT不同类型的创新产品,从药理药效、组织分布与生物分析方法开发、毒理学评价等方向,不断探索研究和实践,为创新的CGT产品提供全面的非临床评价服务,在国内承担CGT产品非临床评价项目的实验室中,昭衍新药保持行业领先。
2022Q1,昭衍新药新签订单有1亿左右是细胞与基因治疗(CGT)订单;2022年上半年,公司承接的CGT订单同比实现翻倍增长;2022年前三季度CGT订单则超过了3亿元,超过了去年全年。
特别值得注意的是,昭衍新药完成了全球首个上市的基因治疗项目SBN-1(ADV-P53)的系统评价,是国内第一个承担基因治疗药物非临床安全性评价研究的机构。
在CDMO方面,2018年成立北京昭衍生物技术有限公司(“昭衍生物”),将以中美两地研发生产基地为依托,为全球创新药研发机构提供可开发性研究、工艺放大优化、质量研究、中试及商业化生产一站式解决方案,是业内唯一能给客户提供“中美两地双厂生产”的服务模式。
此外,公司还设有昭衍药检,其主要面向蛋白药物、疫苗、基因与细胞治疗产品等创新药物质量研究与检定的第三方检测机构,为社会提供创新药物质量标准研究,检定方法建立,标准物质制备及鉴定,细胞库、菌毒种库、原液、成品检验检测,生产工艺质量控制关键步骤如病毒灭活与清除验证等相关服务,致力于支持和促进创新药物的研发及产业化进程。
当前基因治疗领域仍充满挑战与荆棘,作为CGT研发与生产领域首屈一指的CRO/CDMO公司,昭衍新药正在不断地增强自身优势、提高技术壁垒、扩大产能建设、多维度布局科技创新,助力实现CGT药物研发、生产、合规、上市、流通、商业化。
为此,昭衍新药将于2022年11月22日至12月27日,每周二晚19:00—20:00,6期连场,系统性的向大家分享基因治疗的前沿进展及技术壁垒,共同探讨该领域火热赛道下,基因治疗生产与质控研究、质量与检定服务、药理药效研究、生物分析&DMPK、安全性评价、临床检测等方面内容,欢迎从事本领域的专家学者一同参与讨论!

参考资料
【1】《基因治疗研发和生产外包服务市场研究报告》,弗若斯特沙利文咨询公司,2021年5月
【2】《基因治疗的研究进展》,中国细胞生物学学报,2020年4月
【3】《2022基因治疗行业研究报告》,动脉网、蛋壳研究院,2022年6月
