J Clin Invest:整合甲基化组-转录组分析揭示了婴儿MLL-重排型B细胞急性淋巴母细胞白血病的癌细胞脆弱性
时间:2021-09-19 20:25:48 热度:37.1℃ 作者:网络
B细胞急性淋巴细胞白血病(B-ALL)是最常见的儿童癌症。正如其产前起源所预测的那样,婴儿B-ALL(iB-ALL)表现出异常的DNA沉默突变图谱,这表明表观遗传学机制可能在很大程度上影响了该白血病的发生。本研究整合了69例原发MLL-重排型白血病(MLLr)和非MLL-重排型婴儿B细胞急性淋巴细胞白血病(non-MLLr iB-ALL)的全基因组DNA甲基组和转录组数据,这些患者均根据干扰素-99/06方案进行治疗。iB-ALL甲基组标记显示出大量的与正常造血细胞中增强子和转录控制相关的染色质状态的共有的和特异性的改变。DNA甲基化、基因表达和基因共表达网络分析将MLLr与非MLLr iB-ALL区分开来,并确定在MLLr iB-ALL中AP-1复合体成员FOS和JUN以及RUNX因子的共表达和高表达,这与这些基因低甲基化CpG的显著富集一致。整合甲基组-转录组分析确定了一致的癌细胞脆弱性,揭示了与dmCpG信号相关的明显的iB-ALL特异性基因的表达,并证实了AP-1和RUNX成员的表观遗传控制重塑了MLLr iB-ALL的分子网络。最后,使用MLLr iB-ALL患者来源的异种移植物进行体内和体外实验发现药理学抑制或AP-1功能缺失会严重削弱MLLr白血病细胞的生长,为MLLr iB-ALL的新治疗途径提供了理论依据。
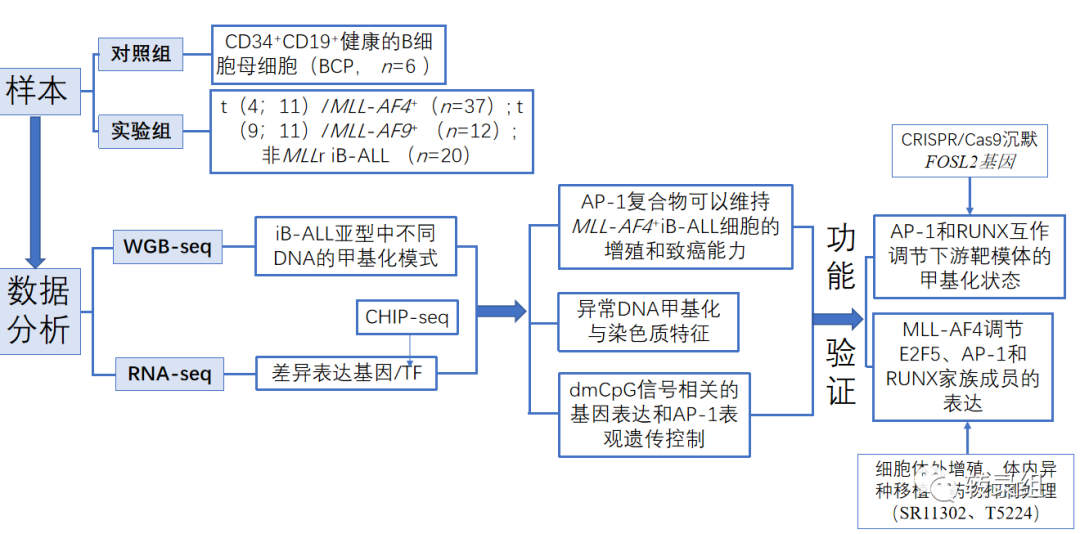
实验设计
结果
1 MLL-AF4+、MLL-AF9+和非MLLr iB-ALL的DNA甲基化图谱
为了描述iB-ALL的整体DNA甲基化状态,研究者首先对高纯度(>95%)的CD19+CD34+iB-ALL白血病母细胞(包含MLL-AF4+(n=2)、MLL-AF9+(n=2)和非MLLr(n=2)这些亚组)和作为对照的2个健康CD34+CD19+B细胞母细胞(BCPs)池中进行了WGB-Seq(补充表1)。无论MLL状态如何,iB-ALL的白血病细胞的特征是DNA甲基化水平总体显著低于健康BCP(图1A和B)。对MLLr iB-ALL中过表达的典型的MLL靶基因FLT3基因的分析,证实了MLLr iB-ALL中靶基因启动子的DNA甲基化特异性缺失,从而验证了WGB-Seq数据(图1C)。接下来,研究者进行了差异甲基化分析,分别在健康BCP和MLL-AF4+、MLL-AF9+、非MLLr iB-ALL患者之间确定了47 713、54 409和50 127个差异甲基化区域(DMRs),大部分为低甲基化区域(图1D和补充表2)。这些差异化甲基区域,尤其是低甲基化位点,富含多个具有DNA重复元素的家族(图1E和补充表3)。WGB-Seq数据通过对n = 69个iB-ALL和n = 6个BCP池的扩展队列中长散布型核苷酸元件(LINE)和DNA甲基化阵列的DNA亚硫酸氢盐焦磷酸测序来进行验证(补充图1A、B以及补充表4)。综合这些数据,iB-ALL的特征是DNA甲基化的全面缺失。
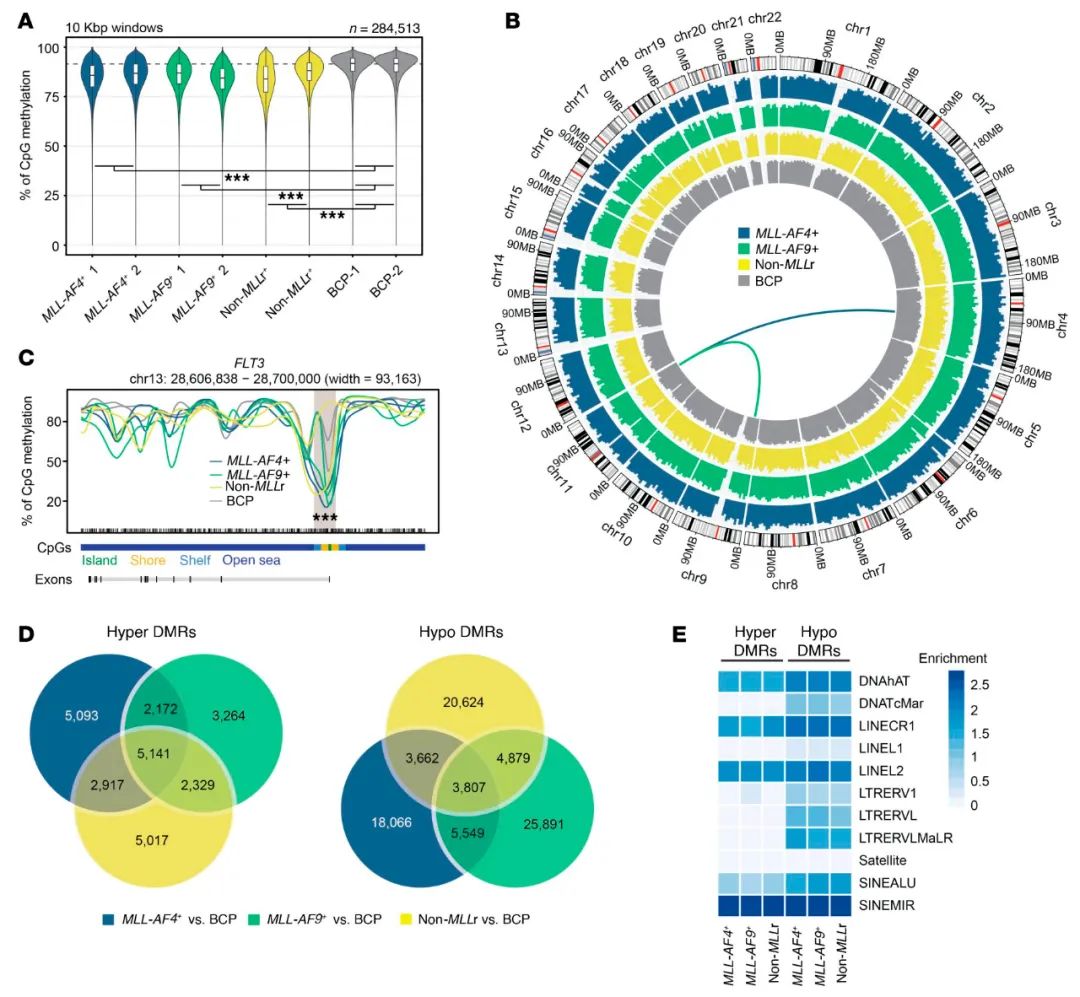 图1.不同iB-ALL亚型的全DNA甲基化状态。(A)小提琴图反映了WGB-Seq鉴定的CpG位点的整体DNA甲基化水平。该图表示在10 Kbp基因组窗口中分割的基因组的CpG甲基化分布百分比。所有iB-ALL亚型的DNA甲基化状态和健康BCP的平均甲基化状态显著不同(***P< 0.001,双侧Wilcoxon秩和检验)。(B)Circos图代表了基因组中DNA甲基化水平。CpG甲基化在平均分布在10 Mbp基因组窗口中,每个iB-ALL亚型的平均DNA甲基化值表示为直方图轨迹。内线识别MLL-AF4+(蓝色)和MLL-AF9+(绿色)移位事件。(C)描绘FLT3基因DNA甲基化图谱的线形图。如下方面板中所指示的,CpG背景和CpG位点位置映射到相应的基因组坐标。MLL-AF4+/MLL-AF9+和非MLLr iB-ALL或健康BCP组之间甲基化差异显著(q<<0.05)的区域用阴影表示,***P<0.001。(D)维恩图表示每个对应的比较中具有一致超甲基化或低甲基化变化的DMR总数。(E)热图显示富集显的超甲基化或低甲基化DMR在不同DNA重复区的log2的优势比(q<0.05)。
图1.不同iB-ALL亚型的全DNA甲基化状态。(A)小提琴图反映了WGB-Seq鉴定的CpG位点的整体DNA甲基化水平。该图表示在10 Kbp基因组窗口中分割的基因组的CpG甲基化分布百分比。所有iB-ALL亚型的DNA甲基化状态和健康BCP的平均甲基化状态显著不同(***P< 0.001,双侧Wilcoxon秩和检验)。(B)Circos图代表了基因组中DNA甲基化水平。CpG甲基化在平均分布在10 Mbp基因组窗口中,每个iB-ALL亚型的平均DNA甲基化值表示为直方图轨迹。内线识别MLL-AF4+(蓝色)和MLL-AF9+(绿色)移位事件。(C)描绘FLT3基因DNA甲基化图谱的线形图。如下方面板中所指示的,CpG背景和CpG位点位置映射到相应的基因组坐标。MLL-AF4+/MLL-AF9+和非MLLr iB-ALL或健康BCP组之间甲基化差异显著(q<<0.05)的区域用阴影表示,***P<0.001。(D)维恩图表示每个对应的比较中具有一致超甲基化或低甲基化变化的DMR总数。(E)热图显示富集显的超甲基化或低甲基化DMR在不同DNA重复区的log2的优势比(q<0.05)。
为了了解iB-ALL中特定遗传特征的潜在机制,研究者对给定iB-ALL亚型的DMR的“特异性”或至少2个iB-ALL亚型的“共有”编码信息进行了超几何模体富集优化(HOMER)分析(补充图2和补充表5)。对低甲基化DMRs的分析显示,iB-ALL的pU.1、EBF、SpiB和ETS显著富集,且与MLL状态无关,这表明这些调节因子可能触发共有的转录白血病发生程序或调节正常B细胞分化。相反,在MLLr iB-ALL中,低甲基化DMRs在AP-1家族转录因子(TFs)FOS和JUN以及RUNX家族成员中有明显的富集。
2 依据基因组位置和CpG背景确定iB-ALL亚型中的不同DNA甲基化模式
为了进一步获得iB-ALL的表观遗传异质性,研究者使用高含量DNA甲基化阵列研究了37个MLL-AF4+、12个MLL-AF9+和20个非MLLr iB-ALL的更大的群体。6个健康的BCP和原始B细胞作为正常的B细胞对应物进行分析,以说明在B细胞分化过程中自然发生的DNA甲基化变化(图2A和补充表4)。重要的是,DNA甲基化阵列和WGB-Seq显示出非常稳健的相关性(补充图3A和B)。无监督主成分分析(PCA)将iB-ALL甲基组与BCP/原始B细胞甲基组区分开(图2B),10000个最可变CpG位点的分层聚类区分了iB-ALL亚型,包括MLL-AF4+和MLL-AF9+亚型,这表明特定的MLL融合可能会差异性地重塑iB-ALL的甲基组图谱(图2C)。
接下来,研究者进行了一项差异甲基化分析,将iB-ALL和健康原始B细胞与健康BCP进行比较,发现在iB-ALL和原始B细胞中分别存在77 596和35 157个差异甲基化CpG(dmCpG)(图2D)。有趣的是,在原始B细胞中观察到的53%(18 470)的dmCpG与iB-ALL样本相同,表明它们代表了B细胞分化过程中自然发生的甲基化变化,因此从iB-ALL特异性dmCpG中去除后,得到了iB-ALL特异性的dmCpG信号(图2D和补充表6)。与WGB-seq序列数据一致,在大多数比较中,低甲基化CpG远远超过了超甲基化CpG(图2D)。但是,在MLLr iB-ALL中,超甲基化CpG依然大量存在。
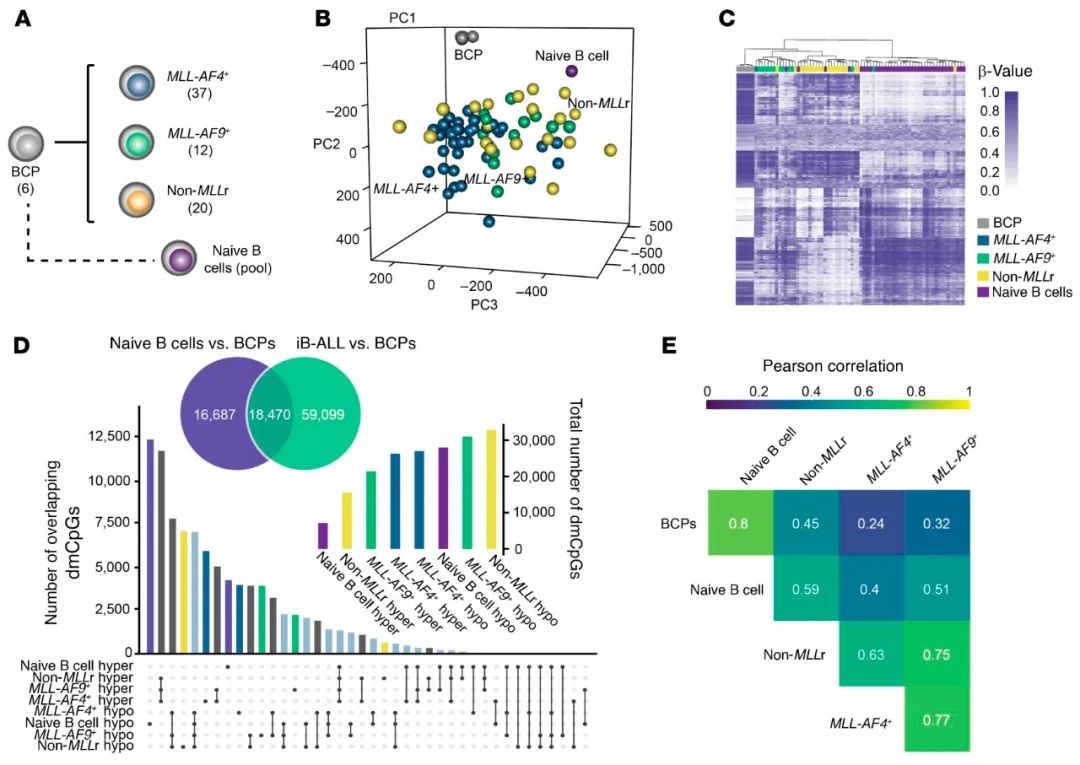
图2.iB-ALL亚型中甲基化差异位点的鉴定。(A)示意图显示了使用人类甲基化EPIC微阵列平台分析的样本数量。(B)关于DNA甲基化研究中的所有样本的758932个CpG位点PCA分析。(C)热图代表了在不同iB-ALL样本,BCP样本,和原始B细胞中(x轴) 10000个最可变CpG位点的甲基化状态(y轴)。(D)条形图显示在所示比较中观察到的共有的(黑色)和特异性的(有色)显著超甲基化或低甲基化CpG位点的数量(FDR<0.05,平均β差异>0.25)。韦恩图表示原始B细胞和iB-ALL之间重叠的dmCpG数量(β>0.25)。dmCpG重叠的原始B细胞和iB-ALL样本被去除后用于下游分析,因为它们代表B细胞分化过程中自然发生的甲基化变化。插图显示了在每种情况下观察到的超甲基化和低甲基化CpG位点的总数。(E)成对皮尔逊相关分析表明不同样本组之间的相似程度。在任何iB-ALL与BCP条件下观察到的共77596个dmCpG用于适当的比较,所有比较均具有统计学意义(P<0.001)。
为了确定不同iB-ALL亚型之间的总体相关性,使用上述iB-ALL相关的77 596个dmCpG进行了一项全局成对相关分析。在这些异常位点中,MLL-AF4+和非MLLr白血病是相关性最低的白血病亚型(皮尔逊相关系数0.63;图2E)。然而,尽管MLL-AF9+白血病与MLL-AF4+样本具有较高的相关性(相关系数,0.77),但它们也与非MLLr样本具有多重变异(皮尔逊相关系数,0.75),这表明MLL-AF9+亚型显示出与MLL-AF4+和非MLLr的重叠的特征。对dmCpG位点的在基因组中分布的详细检测显示,低甲基化CpG在open sea位置和内含子区域富集,而超甲基化CpG在CpG岛、CpG shore和启动子区域富集,无论iB-ALL亚型如何(图3A和B;统计数据见补充表7)。值得注意的是,CpG岛和启动子区的超甲基化CpG在MLL-AF4+iB-ALL中比在MLL-AF9+和非MLLr iB-ALL亚组中更富集,说明了MLL-AF4+iB-ALL的独特的甲基化模式。健康的原始B细胞在CpG岛或启动子区域均未显示出超甲基化CpG的大量富集(图3A和B),这表明在iB-ALL中观察到的dmCpG背景/位置不同于正常B细胞分化的。
接下来,进行了转录因子结合位点(TFBS)富集分析,以预测dmCpG中富集的潜在的参与的转录因子。研究者使用了基因转录调控数据库(GTRD),其中包括迄今为止最完整的统一处理的转录因子的CHIP-seq数据。与WGB-Seq数据一致,对低甲基化CpG的分析显示AP-1家族的转录因子FOS和JUN在MLL-AF4+iB-ALL中显著差异富集(图3C和补充表8),表明它们代表MLL-AF4+中特异性的低甲基化转录因子结合位点。
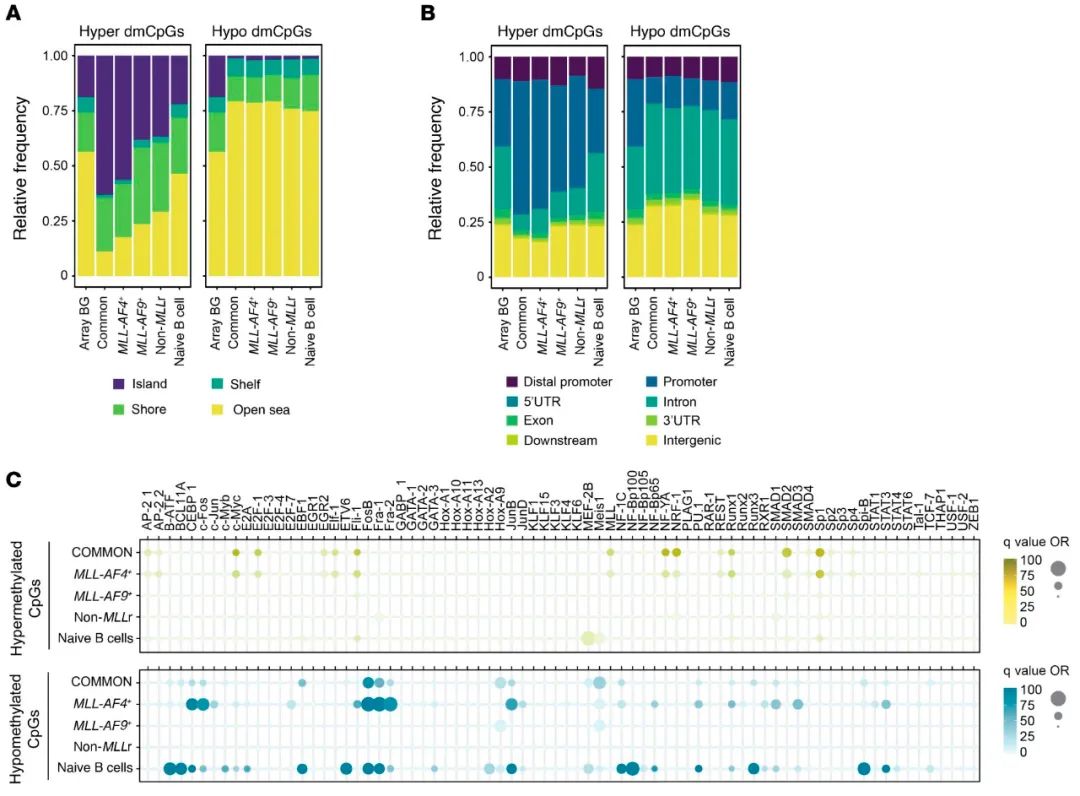 图3.依据基因组定位和CpG背景分析iB-ALL亚型不同的DNA甲基化模式。(A,B)堆叠条形图,描绘了显著的超甲基化或低甲基化CpG相对于其CpG背景(A)或CpG位置(B)的相对频率。(C)气泡图显示根据从GTRD数据库获得的信息确定的对应条件下TFBS的富集情况。顶部(黄色气泡)和底部(蓝色气泡)图分别表示富含超甲基化和低甲基化CpG的TFBS。气泡颜色表示统计显著性(–log2调整后的P值)。点大小表示与EPIC平台的背景分布相比,特定TFBS数据集的log2或富集度。
图3.依据基因组定位和CpG背景分析iB-ALL亚型不同的DNA甲基化模式。(A,B)堆叠条形图,描绘了显著的超甲基化或低甲基化CpG相对于其CpG背景(A)或CpG位置(B)的相对频率。(C)气泡图显示根据从GTRD数据库获得的信息确定的对应条件下TFBS的富集情况。顶部(黄色气泡)和底部(蓝色气泡)图分别表示富含超甲基化和低甲基化CpG的TFBS。气泡颜色表示统计显著性(–log2调整后的P值)。点大小表示与EPIC平台的背景分布相比,特定TFBS数据集的log2或富集度。
3 iB-ALL中异常的DNA甲基化与独特的染色质特征库相关
为了揭示iB-ALL中异常DNA甲基化的潜在功能含义,研究者使用6个公开的组蛋白数据集(H3K4me1,H3K4me3,H3K27ac,H3K27me3,H3K36me3,H3K9me3)进行了全面的区域集富集分析,这些数据集包括来自Roadmap和ENCODE表观基因组联盟的总共5个参考B系造血细胞的表观基因组。研究者发现在H3K4me3标记上,iB-ALL特异性的超甲基化CpG修饰具有差异富集,但正常B细胞特异性的超甲基化CpG修饰没有显著富集(图4A和补充表9)。这在MLL-AF4+ iB-ALL中更为明显。对于所分析的任何组蛋白标记,低甲基化CpG在iB-ALL中都没有差异富集(图4A)。
鉴于组蛋白编码的复杂性、其广泛分布以及某些组蛋白标记可能存在共定位,接下来基于染色质分割数据进行了富集分析,以便于解释这些白血病改变在正常组织中的潜在作用。这种类型的分析整合了来自上述组织的组蛋白标记信息,以便将基因组分成功能相关的染色质状态。超甲基化和低甲基化CpG位点的功能分布在iB-ALL亚组和正常分化的B细胞之间不同(图4B和补充表10)。在iB-ALL中,超甲基化CpG位点在与侧翼和二价转录起始位点(TSS)相关的基因组位点显著富集。这些富集对于MLL-AF4+特异性的超甲基化CpG更为一致。相反,与正常B细胞分化相关的增强子区域的DNA超甲基化富集在iB-ALL中丢失,表明这些调控元件的精确控制在iB-ALL中受损,无论MLL状态如何。在MLLr iB-ALL中,低甲基化的CpG位点也在与被抑制的多梳元件相关的基因组位点显著富集(图4B)。此外,在iB-ALL亚组之间观察到增强子区域的DNA低甲基化差异富集(图4B)。
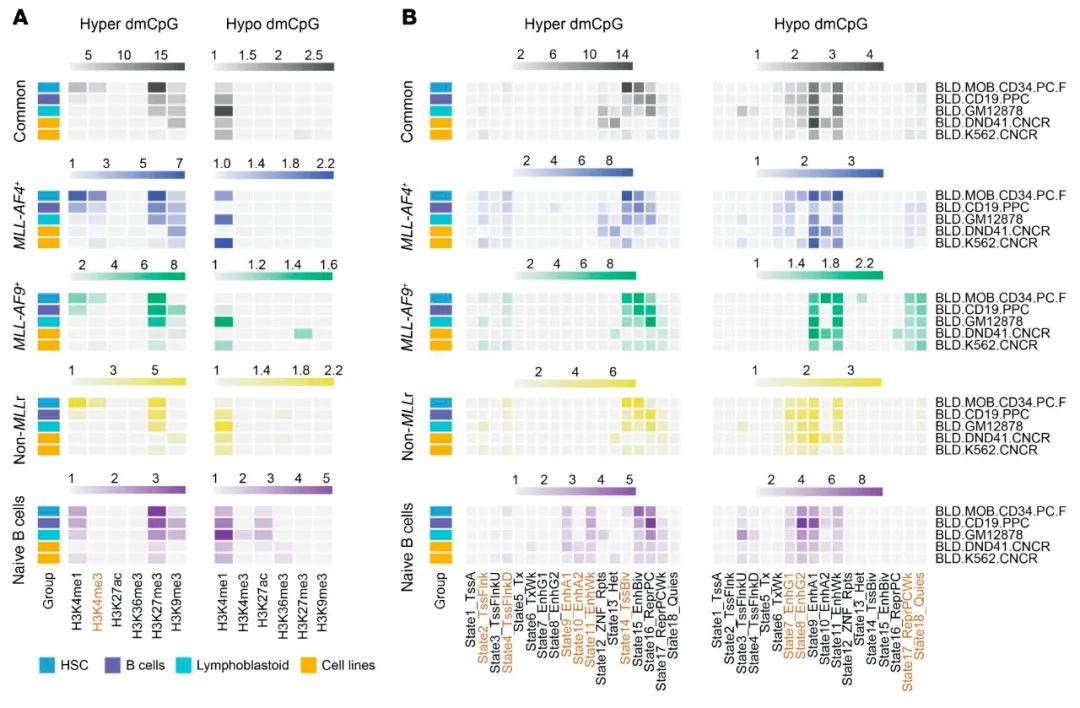
图4.iB-ALL中DNA甲基化的缺失与特定染色体状态的改变有关。(A)热图显示了对原始B细胞、每个iB-ALL亚型特有的或与健康BCP相比至少有2个iB-ALL亚型共有的超甲基化和低甲基化CpG的组蛋白标记富集分析。色标表示先前分析中从NIH Roadmap表观基因组联盟获得的6种常见组蛋白修饰与人类甲基化平台背景分布相比获得的显著dmCpG的优势比。图例表示用于比较的B系造血数据集的类型。(B)热图表示上述条件下超甲基化和低甲基化CpG的染色质状态富集分析。颜色范围表示从NIH Roadmap表观基因组联盟获得的18个染色质状态中观察到的显著dmCpG的优势比。
由于增强子序列的重塑在B细胞身份的建立中起着关键作用,研究者假设异常的增强子甲基化可能作为决定iB-ALL分化状态/阻断的代理。使用来自人类甲基化EPIC平台的原始B细胞特异性的361 dmCpG探针(与增强子图谱中的B细胞特异性增强子重叠)对iB-ALL患者进行分层聚类,将MLL-AF4+与MLL-AF9+和非MLLr iB-ALL患者进行区分。值得注意的是,当将不同iB-ALL组的平均DNA甲基化状态与健康原始B细胞进行比较时,MLL-AF4+和MLL-AF9+iB-ALL显示出比非MLLr-iB-ALL更低的相关性,这表明MLLr iB-ALL本质上由分化程度较低的B细胞表型定义。总的来说,异常的DNA甲基化似乎在功能上与不同iB-ALL亚型的表观遗传重建所涉及的不同分子机制有关。
4 MLLr iB-ALL的转录程序由AP-1复合体的成员控制
iB-ALL中异常的DNA甲基化与转录控制相关的染色质状态的功能有关(图3B)。为了确定显示转录变化的相关分子途径,接下来对iB-ALL患者(n=40)和健康BCP(n=5)进行全面的转录组学分析,从中可以获得相对应的转录组数据的DNA甲基化数据(图5A)。RNA转录数据的无监督PCA分析很容易将iB-ALL患者与健康的原始B细胞和BCP区分开来(图5B)。基于DNA甲基化数据的分层聚类比基于基因表达的聚类更为同质,特别是对于MLL-AF4+iB-ALL(图5C),因为基因表达聚类在MLL-AF4+患者中又分为2个独立的亚组。然而,DNA甲基化和基因表达都可以将非MLLr与MLLr患者分开,这表明非MLLr和MLL AF4+iB-ALL的甲基化和转录组图谱都受到不同的调控。
接下来,对MLL-AF4+、MLL-AF9+、非MLLr iB-ALL和原始B细胞进行了差异表达分析(补充表12)。通路分析分别鉴定了5170个和3919个iB-ALL和原始B细胞与BCP相比的特异性差异表达基因(DEG)(图5D)。MLL-AF4+iB-ALL中基因的表达表现出更大的变异性(图5E),与分层聚类分析中观察到的基因表达异质性一致(图5C),表明MLL AF4+iB-ALL患者具有不同的分子亚群。为了探究iB-ALL转录重组的分子网络,使用RNA序列中663个最可变的基因进行了基因共表达网络分析,发现了3个基本的基因差异表达模块(图5F和补充图5A)。模块1和模块2富集在MLL-AF4+和MLL-AF9+亚群中,在健康的原始B细胞和BCP中几乎没有表达,而模块3主要将MLL-AF4+iB-ALL与MLL-AF9+和非MLLr iB-ALL区分开(图5F)。对这些模块的详细分析显示,模块1、2和3分别富集在细胞激活、炎症反应和细胞分裂相关的基因本体类别的不同基因集上(补充图5B、C)。此外,模块2显示与NF-κB、p53和STAT信号通路相关的基因集显著富集,而模块3显示与G2M检查点、E2F靶点和有丝分裂纺锤体相关的基因集富集。每个模块中共表达了不同的转录因子(补充图5D、F)。重要的是,模块2,最能将MLLr iB-ALL与非MLLr iB-ALL、正常原始B细胞和BCP区分开来,显示出AP-1复合物中转录因子FOS和JUN一致的高表达(补充表5E),与MLLr iB-ALL的FOS和JUN中低甲基化CpG的显著富集一致,表明这些AP-1成员的表观遗传控制可能对MLLr iB-ALL的发病进程有影响。
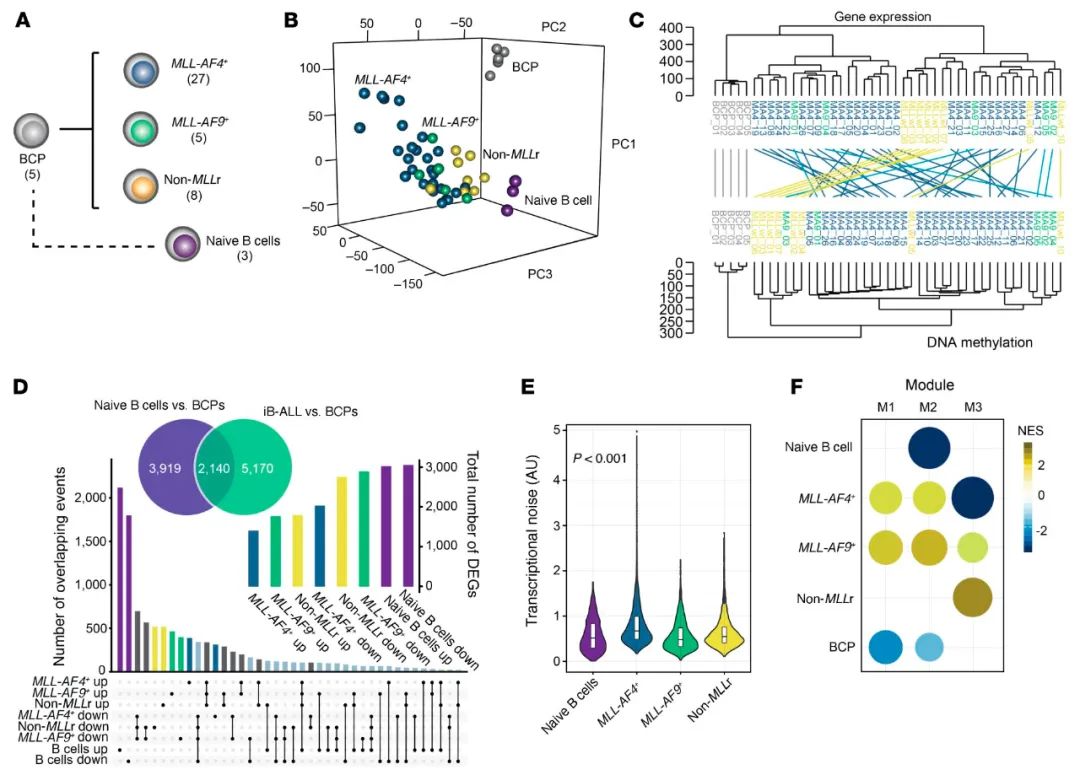
图5.基因共表达网络分析揭示了AP-1复合体成员JUN和FOS在MLLr iB-ALL中可能是潜在的致病因素。(A)描述RNA-Seq分析的样本数量的示意图。(B)通过RNA-Seq分析的所有样本中的基因表达矩阵(每千个碱基的转录每百万映射读取的碎片[FPKM]值)生成的主成分分析。(C)成对DNA甲基化和基因表达数据之间的聚类关系的缠结图表示(缠结=0.31,共表相关分数=0.4)。(D)条形图显示了与健康BCP进行每次比较时观察到的显著上调或下调的共有的(黑色)和特异性的(有色)基因的数量。内部图形表示每种情况下的差异基因总数。维恩图表示与健康BCP相比,原始B细胞和iB-ALL之间重叠的DEG数量。P<0.001,单端超几何检验。(E)小提琴图显示了与健康BCP相比,每种情况下DEGs的基因表达变异系数(log2倍变化)。P<0.001,所有比较均采用双侧Wilcoxon秩和检验。(F)散点图,描述各组中特定基因模块的标准化富集分数(NES)和贡献(由点大小表示)。
5 综合分析揭示了MLLr iB-ALL中与dmCpG信号相关的基因表达和AP-1成员的表观遗传控制
研究者使用ELMER在iB-ALL中整合相对应的DNA甲基化和RNA-Seq数据。ELMER相关性是使用iB-ALL亚型的特定的dmCpG或iB-ALL中共有的dmCpG(大于2个iB-ALL亚组共有的)以及RNA-seq中所有基因的表达来计算的(图6A)。在共有的、MLL-AF4+、MLL-AF9+或非MLLr中,dmCpG与基因表达呈强相关性,分别高达10%、17%、33%和25%(补充表13)。为了便于数据解释,重点研究了有表观遗传调控经典模型支持的相关性,发现DNA甲基化与基因表达密切相关:超甲基化变化与基因抑制相关,而低甲基化变化与基因激活相关(图6B和C)。基因表达相关的dmCpG探针在CpG岛和远端启动子处富集,但在open sea位置和基因间区域的表达显著不足(补充图6A和B;补充表7)。此外,这些CpG位点的分布与组蛋白标记和与活性转录相关的染色质状态有关(补充图6C和D以及补充表14和15),确定了iB-ALL中与基因表达相关的dmCpG。例如,iB-ALL中CpG探针cg07893009的低甲基化与LMO2基因的增加相关(图6D),而CpG探针cg06365535的超甲基化与iB-ALL中BRIP1基因的抑制显著相关(图6E)。此外,使用全基因表达相关dmCpG探针进行的反应体途径富集分析显示,MLLr iB-ALL中的基因抑制和超甲基化CpG之间存在联系,与细胞周期、有丝分裂、DNA复制和修复有关的细胞途径受损有关(图6F)。
然后,研究者试图确定主要转录因子对基因表达互作的dmCpG的作用。这些dmCpG的TFBS的富集是使用ELMER和来自HOCOMOCO数据库的公开可用的人类的结合模型进行的。在SP和E2F转录因子中发现在超甲基化CpG中的显著富集(图6G)。然而,引人注目的是,许多FOS/JUN成员和RUNX蛋白在低甲基化CpG中大量富集,特别是在MLL-AF4+iB-ALL患者中,进一步表明AP-1成员的表观遗传控制重塑了MLL-AF4+iB-ALL的分子网络。
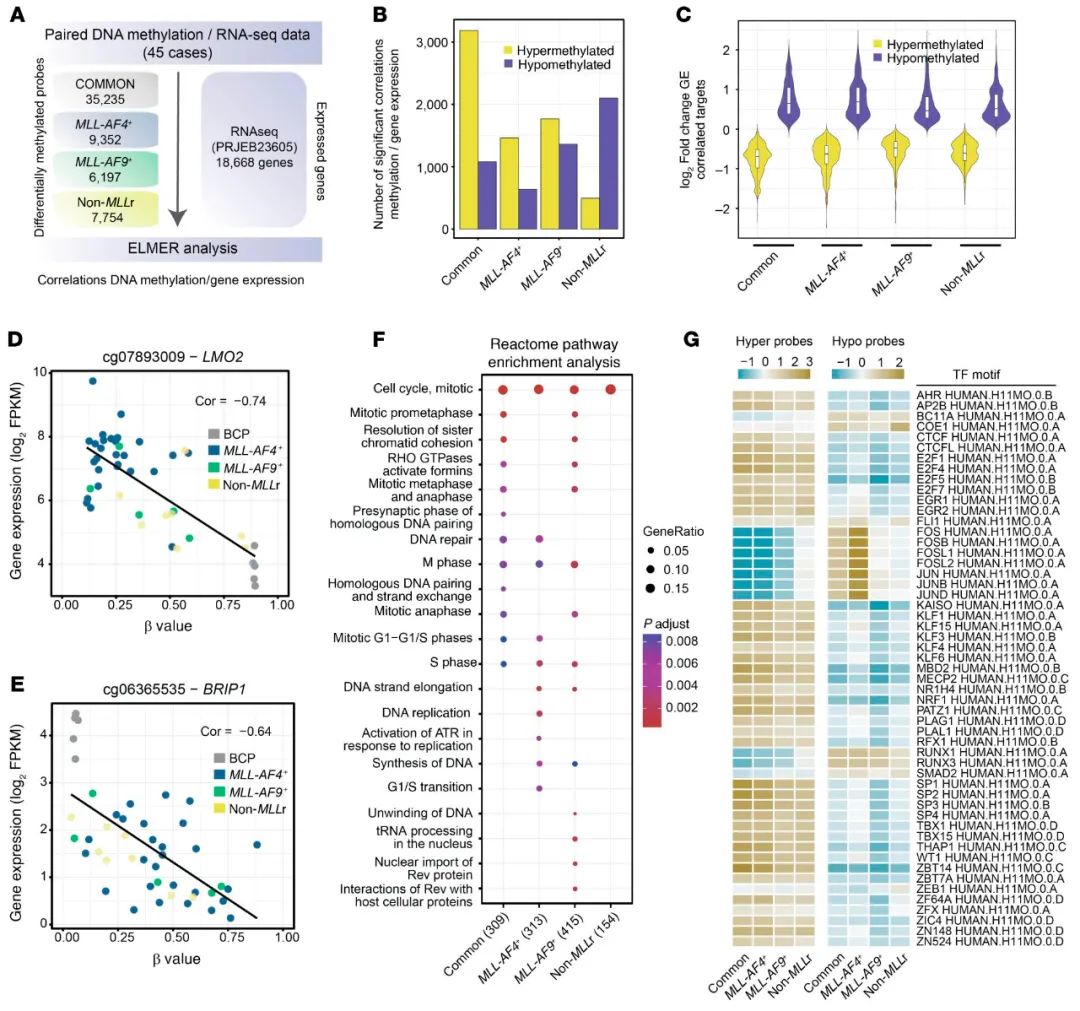
图6.整合DNA甲基化和基因表达数据。(A)描述使用ELMER算法整合DNA甲基化和RNA序列数据的工作流程的示意图。(B)描述与超甲基化或低甲基化CpG相关的基因表达数量的条形图(皮尔逊相关系数>0.5)。(C)小提琴图显示与dmCpG探针一致相关的基因的基因表达变化分布(指示组与健康BCP的log2倍数变化)。(D,E)散点图,分别显示基因LMO2(D)和BRIP1(E)的DNA甲基化和基因表达之间的相关性。皮尔逊相关(Cor)分数表示每次比较。(F)反应途径富集分析散点图。与背景数据集相比,与DNA甲基化相关的基因用于富集计算,背景数据集包括我们RNA-Seq数据集中所有可检测表达的基因(18 668)。颜色范围表示所表示本体的重要性(调整后的P值),而点大小表示所识别的点击数与给定本体中点击总数之间的比率。(G)基因表达中TFBSs富集的热图表示——与超甲基化或低甲基化CpG相关。颜色范围表示通过ELMER算法计算的给定图案(对数优势比)的丰富度或不足。
6 AP-1-和RUNX-互作因子通过调节下游靶模体的甲基化状态重塑iB-ALL的调节网络
接下来,研究者探讨了主要转录因子是否在表观遗传学上控制iB-ALL中的基因表达。研究者首先绘制了E2F5(一种富含超甲基化CpG位点的转录因子)的基因组背景图(图6G),并在E2F5 TSS上游1 Mbp范围内发现了11个高度相关的CpG探针(图7A)。该基因组区域在iB-ALL中特异性超甲基化(图7B),E2F5的表达受到抑制,特别是在MLL-AF4+iB-ALL中(图7C)。重要的是,在MLL-AF4+iB ALL中E2F5模体的平均DNA甲基化显著增加(图7D),表明E2F5的缺失促进了这些特定位点的甲基化。此外,在所有iB-ALL组中,E2F5靶点的表达均低于预期水平(图7E)。正如预期的那样,E2F5靶点的表达在原始B细胞中也受到抑制,因为E2F5是最终分化成熟的B细胞中的一种转录抑制因子。
然后重点研究了AP-1成员,它们在MLL-AF4+iB-ALL的低甲基化CpG位点高度富集(图6G)。发现了5个高度相关的增强子连接的CpG探针分布在FOLS2启动子下游0.35 Mbp处(图7F)。这些dmCpG基因座在iB-ALL中被强烈地去甲基化,特别是在MLL-AF4+患者中(图7G)。这种dmCpG的低甲基化伴随着iB-ALL中FOSL2表达的显著增加(图7H)。因此,iB-ALL中结合模体的平均DNA甲基化显著降低,特别是在MLL-AF4+患者中(图7I),并且iB-ALL中FOSL2靶蛋白的表达比原始B细胞和在iB-ALL中的偶然预期高(图7J)。
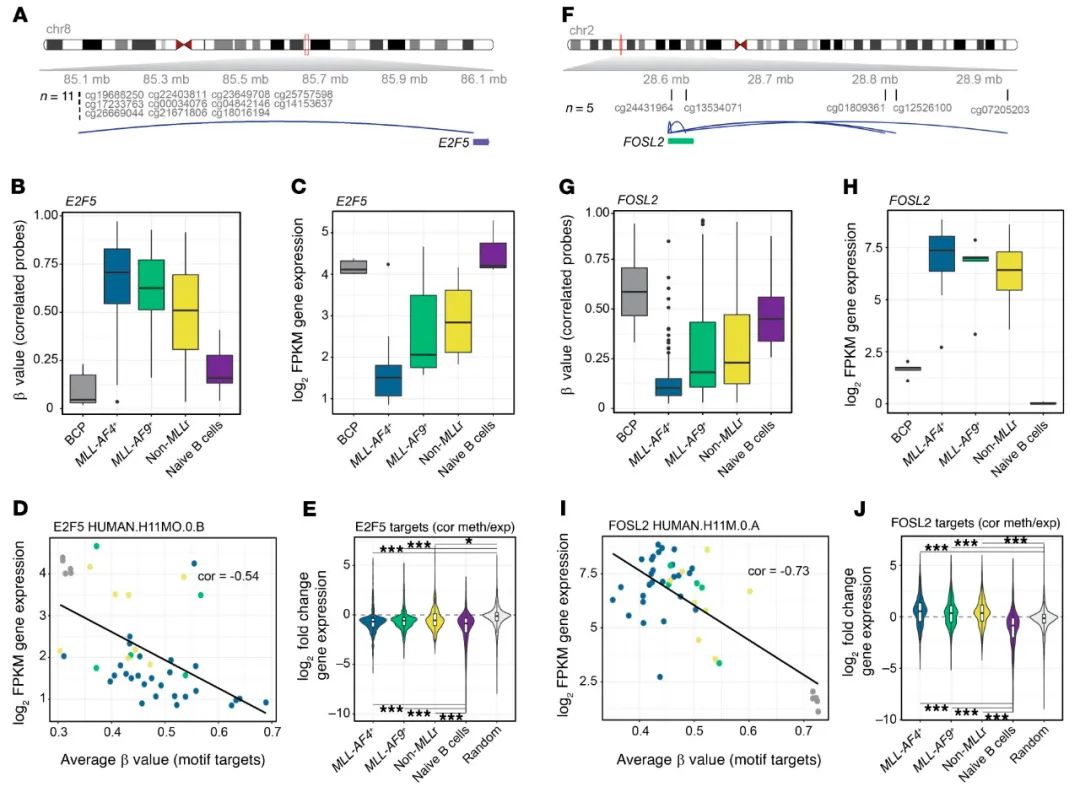
图7.E2F和AP-1互作因子控制了下游靶模体的甲基化状态。(A)表意文字代表与dmCpG位点相关的E2F5表达的基因组位置。n表示使用ELMER算法识别的相关dmCpG的数量。(B)箱线图描绘了不同组间重要的与CpG探针相关的E2F5表达的平均甲基化。(C)反映所示组中E2F5表达的箱线图。(D)散点图显示E2F5模体靶点的平均DNA甲基化与E2F5表达之间的相关性。彩色点:蓝色,BCP;红色,MLL-AF4+;绿色,MLL-AF9+;黄色,非MLLr。(E)小提琴图显示了用ELMER算法获得的E2F5模体的目标基因的基因表达变化分布(指示组与健康BCP的log2倍数变化)。*P<0.05,***P<0.001,双侧Wilcoxon秩和检验。所有包含在iB-ALL亚组中的具有E2F5模体的相关基因用于表示B细胞基因表达分布。“随机”组包括对B细胞组中包含的相同数量的基因进行随机取样,但使用原始基因表达矩阵,包括RNA-Seq数据集中可检测表达的所有基因。(F-J)与A-E相同,但适用于FOSL2。
为了证实FOSL2在重塑iB-ALL的表观遗传图谱中发挥作用,进行了更深入的网络分析,以验证FOSL2靶基因表达与高度相关的CpG之间的联系。顶部网络节点的特征是低甲基化FOSL2结合模体的存在和相应FOSL2靶点的上调(补充图7A)。进一步研究了FOSL2调节网络的两个节点,DUSP10和CD44,它们是iB-ALL中高度表达的致癌基因。DUSP10和CD44相关的dmCpG显著低甲基化(补充图7B和C),DUSP10/CD44在iB-ALL中过量表达,特别是在MLLr iB-ALL中(补充图7D和E)。
为了证明FOSL2上调对MLL-AF4+白血病甲基化状态的功能影响,研究者在CRISPR-Cas9介导的敲除FOSL2的MLL-AF4+SEM细胞中进行了功能分析(图8A)。总的来说,观察到FOSL2敲除后基因组发生了显著的整体超甲基化(图8B),在FOSL2KO和SEM-WT细胞之间共鉴定出12 218个超甲基化和3 018个低甲基化位点(图8C和补充表16)。随后对这些FOSL2调控的dmCpG进行的HOMER模体的富集分析表明,最显著的TFBS主要与AP-1复合体的成员有关(图8D和补充表17)。为了在基因表达水平上进一步验证这些观察结果,在SEM-WT和SEM-FOSL2KO细胞中进行了RNA-Seq。观察到,在FOSL2缺失的情况下,与AP-1通路激活相关的基因显著减少(图8E)。
此外,FOSL2KO细胞中的FOSL2靶点显著下调(图8F)。在MLL-AF4+细胞中FOSL2去除的背景下,以NUDT21和FSCN1基因为例,进一步分析FOSL2相关的dmCpG及其对基因表达的潜在影响(图8G和H)。FOSL2去除后,NUDT21 TSS附近的一个CpG岛低甲基化,导致3′端RNA切割和多聚腺苷酸化因子的表达增加。相反,位于FSCN1 TSS附近的FOSL2模体的DNA超甲基化与SEM细胞中此类基因的下调相关。这些实验验证了在拓扑结构网络方法中观察到的FOSL2的作用。
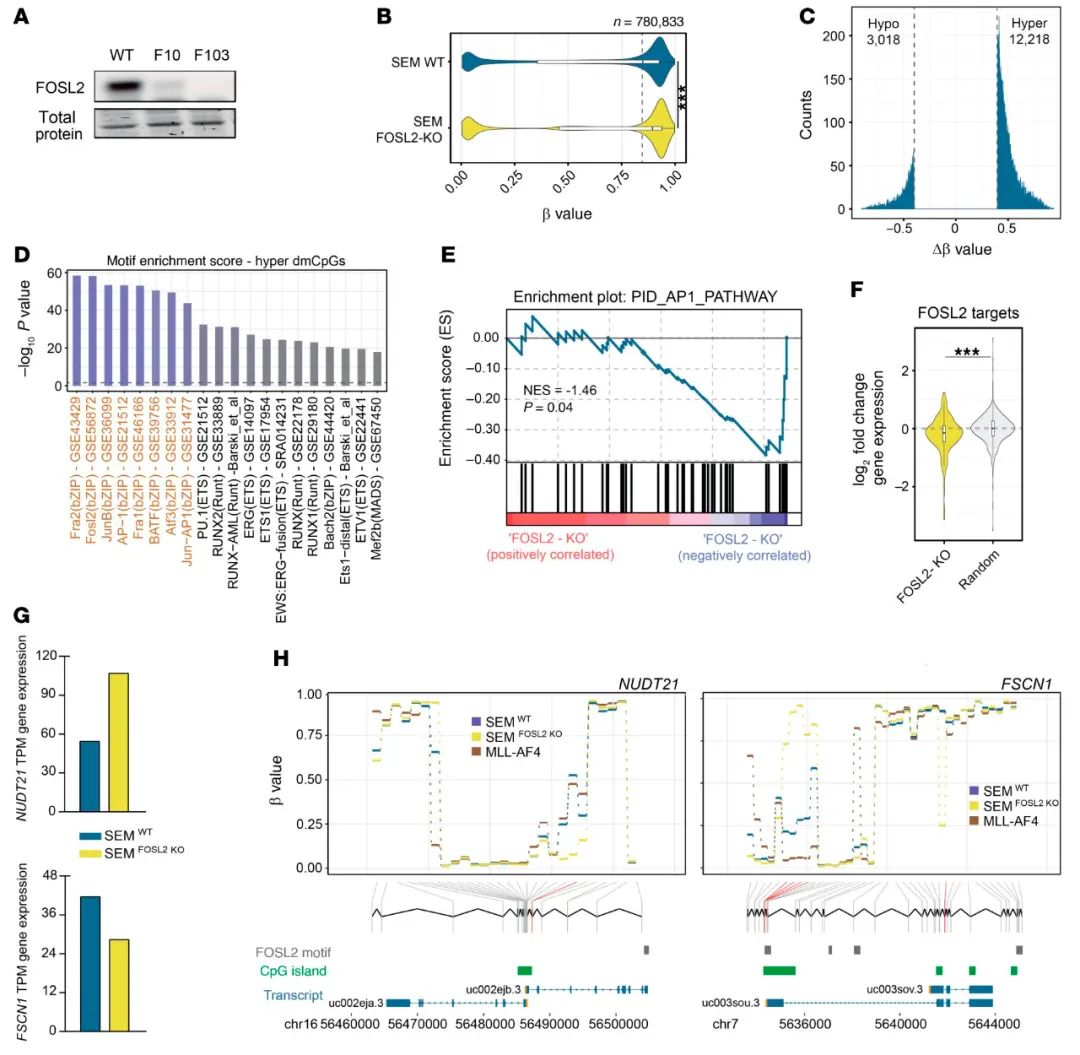
图8.FOSL2调节了MLL-AF4+的甲基组。(A)Western blot证实CRISPR/Cas9编辑的SEM细胞的2个克隆中的FOSL2被敲除。总蛋白是指通过Ponceau染色测定的上样对照。(B)小提琴图显示SEM-WT和SEM-FOSL2KO细胞的平均甲基化情况。***P<0.001,双侧Wilcoxon秩和检验。(C)直方图显示SEM-FOSL2KO和SEM-WT细胞之间的超甲基化和低甲基化CpG位点的数量(平均β差异>0.40)。(D)HOMER模体富集分析强调了在SEM-FOSLKO细胞中超甲基化CpG背景下鉴定的顶级富集TFBS。(E)图示从SEM-FOSL2KO和SEM-WT的基因表达数据以及对应于AP-1途径的基因集获得的富集分析的图。使用GSEA的预排序模式计算NES和显著性得分。(F)小提琴图显示FOSL2靶基因(注释的dmCpG数量>3,n=107)或基因的随机子集(n=500,包括RNA-Seq数据集中可检测表达的所有基因***P<0.001,双侧Wilcoxon秩和检验)的基因表达变化分布(SEM-FOSL2KO与SEM-WT细胞的log2倍数变化)。(G)代表SEM-WT和SEM-FOSL2KO细胞池中NUDT21和FSCN1基因RNA-Seq表达结果的条形图(FPKM)。(H)描述在NUDT21和FSCN1基因附近被询问的甲基化探针的基因组定位和β值得分的图。颜色表示指定组的甲基化情况,包括MLL-AF4+iB-ALL所有患者样本以进行适当比较。显著的dmCpG(平均β差异>0.40)以红色突出显示。下方面板显示上述探针和基因的基因组背景,包括FOSL2模体(GTRD数据库)和CpG岛的位置。TSS以橙色突出显示。
接下来,研究者探索了RUNX1的调控网络,以便将结论扩展到在MLL-AF4+iB-ALL中异常表达的MLL-AF4+的另一个经典分子靶点。研究者发现4个高度相关的增强子连接的CpG位点分布在RUNX1的基因组位点上(补充图8A)。与FOSL2类似,这些dmCpG基因座在iB-ALL中强烈地去甲基化,特别是在MLL-AF4+患者中(补充图8B)。这种dmCpG的低甲基化伴随着RUNX1表达的显著增加(补充图8C)。此外,iB-ALL中RUNX1结合模体的平均DNA甲基化显著降低(补充图8D),iB-ALL中RUNX1靶点的表达偶尔高于原始B细胞和在iB-ALL中的偶然预期(补充图8E)。RUNX1前几位靶基因的网络分析显示,在MLLr iB-ALL中,RUNX同源物RUNX2以类似的方式被调节(补充图8F)。RUNX2相关CpG在MLLr iB-ALL中显著低甲基化(补充图8G),而相反地,在MLLr iB-ALL中RUNX2过度表达(补充图8H)。总的来说,DNA甲基化和基因表达之间的复杂关系通过主要的分子枢纽重塑了iB-ALL的调控网络,如AP-1复合体的成员和RUNX1。
7 MLL-AF4调节E2F5、AP-1和RUNX家族成员的表达
MLLr iB-ALL的基因沉默突变图谱表明MLL-AF4+移位可能足以启动白血病发生。为了探究MLL-AF4自身是否能够激活在iB-ALL患者中观察到的转录程序,研究者分析了Lin及其同事最近的一项研究中的RNA-Seq数据,该研究在人类MLL和小鼠AF4之间建立了白血病嵌合融合。研究者发现E2F5、FOSL2和RUNX1的表达在MLL-AF4转导的CD34+细胞(图9A)和人类原发性MLL-AF4+iB-ALL患者(图7)中受到类似的调节,这表明MLL-AF4在没有进一步的基因组突变的情况下触发类似的转录组重新连接。此外,在AP-1复合体的多个成员中观察到类似的基因激活特征,包括FOS、FOSL1、FOSB、JUN、JUNB和JUND(补充图9A)。值得注意的是,发现CD34+细胞中的MLL-AF4或SEM细胞中的MLLN/AF4C直接结合到FOSL2和RUNX1(图9B)、FOS、FOSL1、JUN、JUNB和JUND的启动子区域(补充图9B),表明MLL-AF4直接调控这些靶基因。
为了在内源性人类移位的基因组背景下验证这些靶点的调节,研究者在CD34+细胞中构建了CRISPR/Cas9介导特定位点位点t(4;11)/MLL-AF4+,和Secker及其同事最近报道的一样。FOSL2和RUNX1的表达(图9C),以及AP-1家族的其他成员,包括FOS、FOSL1、FOSB、JUNB和JUND,在内源性MLL-AF4+移位事件后上调(补充图9C)。接下来,对这些CD34+细胞的t(4:11)/MLL-AF4+移位后的DNA甲基组进行了全面分析,并在移位事件诱导后观察到总共5 011个超甲基化CpG和6 752个低甲基化CpG(图9D和补充表18)。有趣的是,在t(4:11)/MLL-AF4+诱导的CD34+细胞中,80%以上的dmCpG位点位于iB-ALL患者中受MLL-AF4影响的相同甲基化基因上,产生了大约25%的CpG位点的精确位置重叠(图9E)。尽管如此,AP-1成员是经t(4:11)/MLL-AF4+低甲基化位点鉴定的最重要的TFBS之一(图9F和补充表19)。总之,MLL-AF4在没有其他遗传损伤的情况下控制AP-1成员的表达,并与观察到的结合模体的去甲基化直接相关,表明AP-1成员的存在可能保护这些特定位点的DNA甲基化。
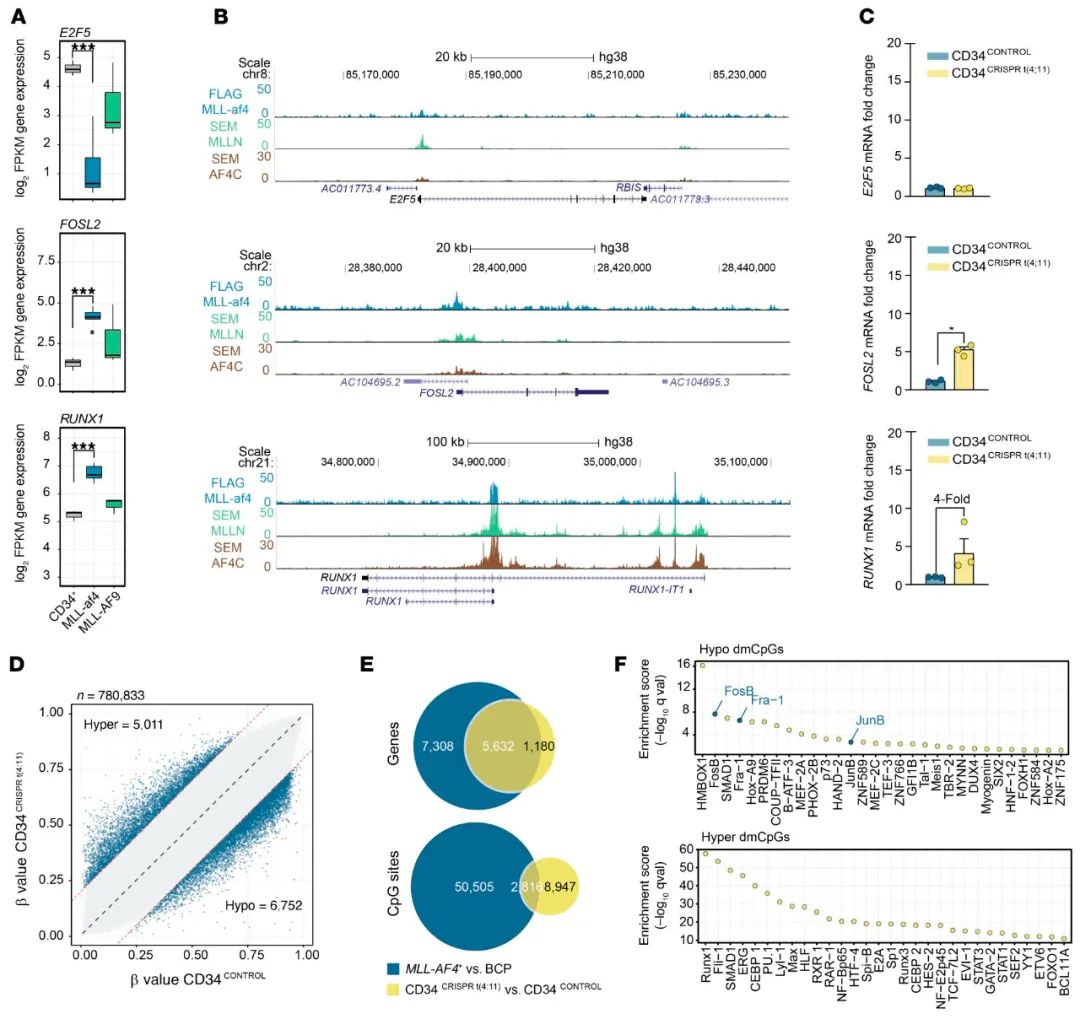
图9.MLL-AF4 调节 AP-1成员和RUNX1的表达,重塑了CD34+细胞的甲基组图谱。(A)箱线图描绘了E2F5、FOSL2和RUNX1基因在健康未转导的CD34+细胞或用人:小鼠嵌合MLL-AF4或人MLL-AF9转导的CD34+细胞中的平均表达。***P<0.001,双侧韦尔奇t检验,n=3;n=6;n=3。(B)UCSC基因组浏览跟踪在E2F5、FOSL2或RUNX1基因附近MLL-AF4(在CD34+细胞中)或MLLN/AF4C(在SEM细胞中)的结合模式。数据分别表示从NCBI数据库基因表达综合[GEO]GSE84116和GSE74812获得的芯片序列信号。(C)显示未编辑的CD34+细胞(CD34对照)和携带位点特异性t(4;11)/MLL-AF4+(CD34CRISPRt(4;11))细胞之间E2F5、FOSL2或RUNX1表达的RT-PCR相对倍数变化的条形图。条形图表示平均值±标准差,*P<0.05,双侧韦尔奇t检验。(D)散点图,指示超甲基化或低甲基化CpG位点在由CRISPR t(4:11)/MLL-AF4+编辑的CD34+细胞上的数量(平均β差异>0.20)。(E)韦恩图表示MLL-AF4+患者和CD34+t(4;11)/MLL-AF4+(CD34CRISPR-t(4;11))细胞,分别与健康的BCP或未经修饰的CD34+细胞进行比较。P<0.001。所有比较均采用单尾超几何检验。(F)根据从GTRD数据库获得的信息,显示CD34+t(4;11)/MLL-AF4+(CD34CRISPR-t(4;11))细胞中低甲基化和超甲基化CpG背景下TFBS富集的图。y轴表示与EPIC平台背景分布相比,特定TFBS数据集的-log10调整P值富集度。
8 AP-1复合物在体外和体内实验中都可以维持MLL-AF4+iB-ALL细胞的增殖和致癌能力
AP-1复合物在MLLr iB-ALL患者中观察到的表观遗传和转录重构中起着核心作用。因此,下一步需要在体外和体内探究靶向AP-1是否代表着MLLR B-ALL的治疗机会。研究者通过FOS显性负效应亚型(dnFOS)(图10A)或CRISPR/Cas9介导的FOSL2基因去除(图8A)的转导,干扰SEM细胞中的AP-1活性。在体外细胞增殖过程中,dnFOS表达被抑制(图10A),dnFOS-SEM细胞的增殖能力降低4倍(图10B)。此外,在体内移植dnFOS-SEM细胞的异种移植物中,肿瘤负担(通过生物发光[BLI]和外周血[PB]细胞流式和脾肿大分析)显著降低(图10C-E)。在体外和体内实验中,通过CRISPR介导的SEM细胞FOSL2基因的消除,在增殖、克隆形成和致癌能力方面获得了更强烈的抑制(图10F-J)。
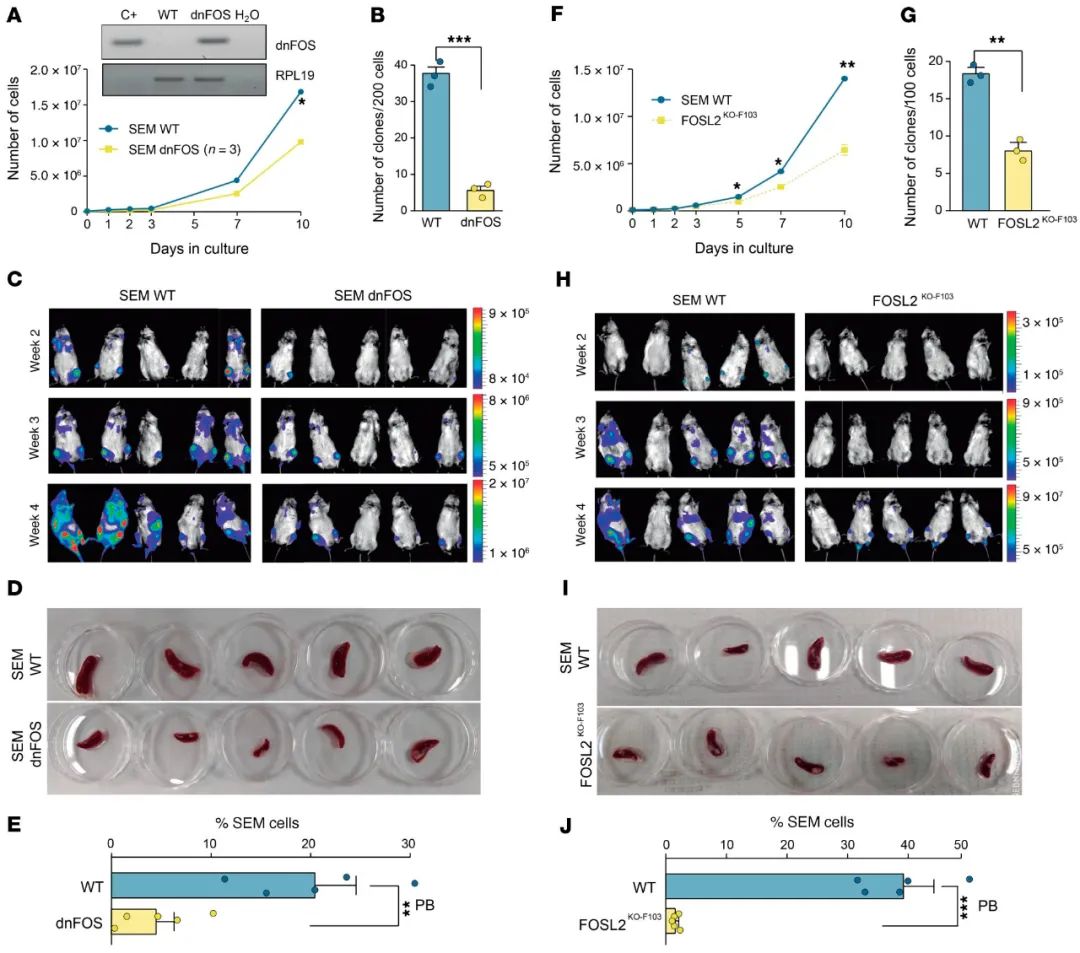
图10.AP1复合体在体内和体外实验中都可以削弱MLL-AF4+ B-ALL细胞的增殖。(A)dnFOS-SEM细胞体外增殖受损。展示了一个具有代表性的实验(n=3)。插入代表RT-PCR,确认转导SEM细胞中dnFOS的表达。RPL19用作加载控件。(B)dnFOS-SEM细胞的克隆形成能力受损。(C)BLI成像显示移植dnFOS-SEM细胞的NSG小鼠体内肿瘤负荷显著降低(n=5只小鼠/组)。(D)移植WT和dnFOS扫描电镜细胞的小鼠脾脏的终点宏观图像。(E)用WT和dnFOS-SEM细胞移植的小鼠外周血的终点白血病负荷。(F-J)与A-E相同,但是关于CRISPR编辑的FOSL2KO-F103-SEM细胞。*P<0.05,**P<0.01,***P<0.001,双侧韦尔奇t检验。条形图表示平均值±标准差。
然后,通过可用的化学抑制剂SR11302或T5224对AP-1进行药理学抑制,并观察到t(4;11)/MLL-AF4+CD34+细胞增殖的削弱随药剂量的增加而增加(图11A)。为了验证观察结果,研究者在一组额外的MLLr和非MLLr白血病细胞系中进行了T5224 AP-1抑制剂的体外药理学分析。值得注意的是,证实了T5224在抑制MLLr(而非非MLLr)细胞增殖方面的特异性和药物剂量依赖性作用(图11B、C)。
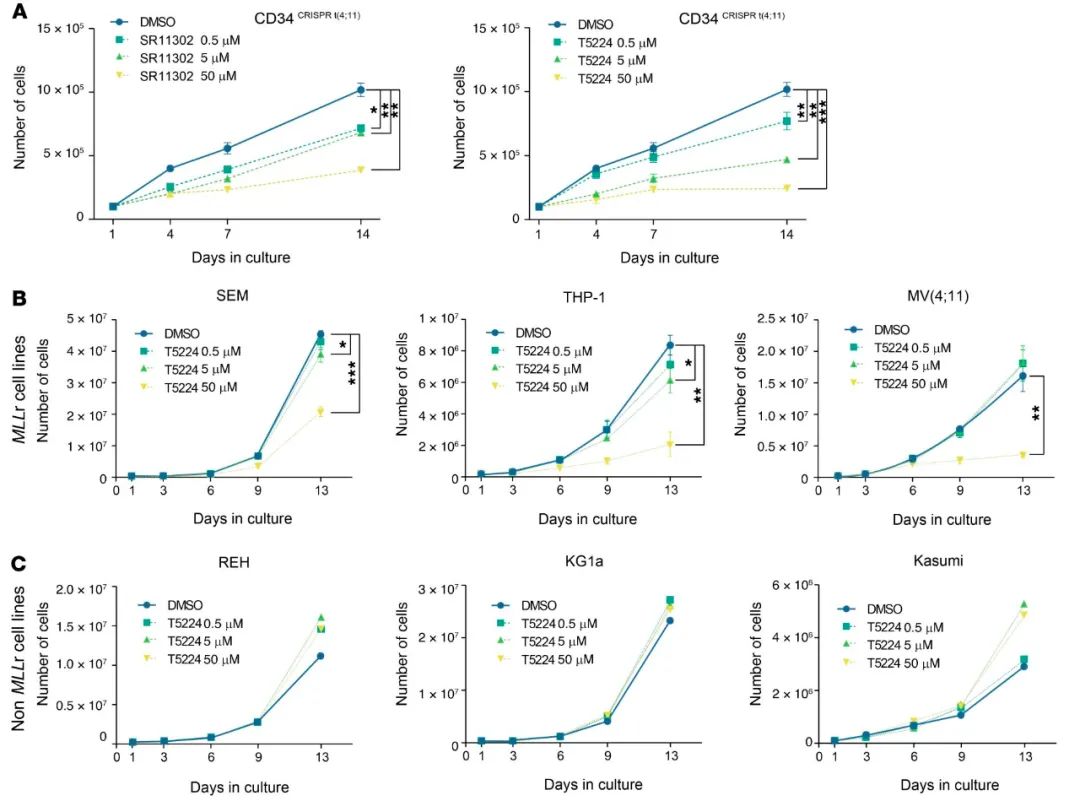
图11.药物抑制 AP-1复合体可以特异性的削弱 MLLr 白血病细胞的增殖。(A)CD34CRISPR t(4;11)用AP-1化学抑制剂SR11302(左)或T5224(右)处理后的细胞。DMSO(二甲基亚砜)条件在两种治疗中都很常见,并且在两种图中都有表示,以便于适当的可视化。(B和C)经增加剂量的T5224抑制剂处理的几种MLLr(B)和非MLLr(C)白血病细胞系的体外增殖。结果P值用Holm方法进行多重比较调整,*P<0.05,**P<0.01,***P<0.001,单侧韦尔奇t检验。线形图表示平均值±标准差(n=3),颜色表示不同药物治疗的指示浓度。
最后,为了验证药理学方法在体内的有效性,将T5224抑制剂与基于长春新碱、地塞米松和L-天冬酰胺酶(VXL治疗)的B-ALL标准治疗相结合,并在实验室建立的临床前的B-ALL PDX模型中应用(图12A)。在BM,脾,和外周血中,测定2个完整VXL周期后的最小残留病灶(MRD)水平和随后的复发率。治疗前确定白血病植入,以便根据BM白血病负荷使小鼠接受VXL或VXL+T5224(图12B)。在两个完整的VXL周期(第20天)后,研究者分别观察了进行VXL治疗的10只MRD阴性小鼠中的8只和VXL+T5224治疗10只MRD阴性小鼠中的9只(%blasts<5%)(图12C)。在终点分析中,T5224介导的治疗有明显改善。在T5224治疗组中,带有BM白血病移植物的小鼠比例较低(30% vs.50%)。同样,在T5224治疗组中,BM(16% vs.34%)、脾脏(26% vs.9%)和外周血(16% vs.3%)的白血病负荷持续较低(图12D)。综上所述,这些结果表明,AP-1复合物的转录因子在MLL AF4+iB-ALL的发病机制和白血病发生中起着核心作用,并可能为MLLr iB-ALL提供一个尚未探索的临床治疗机会。
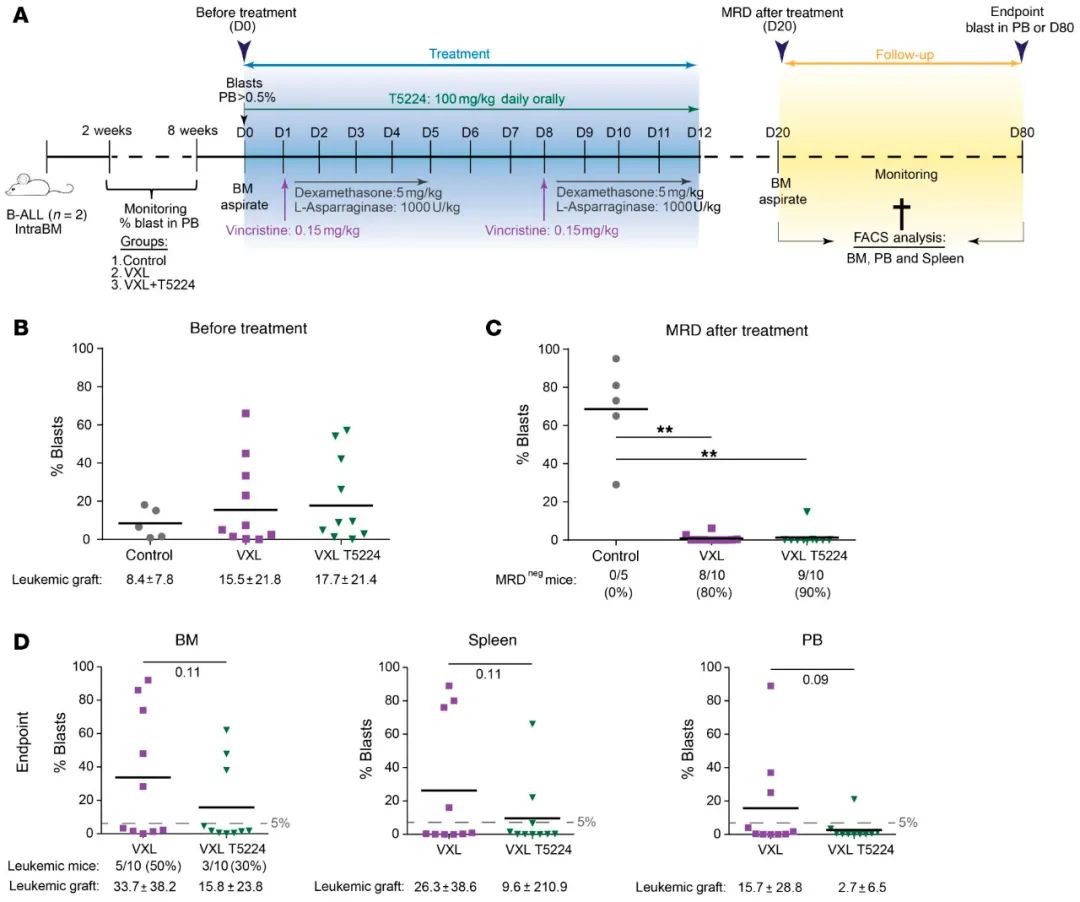 图12.AP-1抑制剂 T5224 与VXL协同作用可以一定程度地减轻活体MLLr iB-ALL异种移植的白血病侵袭性。(A)用VXL单独或与T5224抑制剂联合治疗的临床前MLLr iB-ALL PDX模型的体内实验设计。在完成2个基于VXL的化疗周期后1周(第20天),在BM中评估MRD。监测BM、脾脏和外周血的后续复发情况。(B)在治疗前(第0天)确定BM白血病植入,以便根据BM白血病负荷对小鼠进行同样的随机分组,以接受VXL或VXL+T5224治疗。(C)完成2个VXL±T5224周期后1周(第20天),点图监测BM中的MRD。(D)在随访期结束时,白血病小鼠的比例以及BM、脾脏和外周血中的白血病植入水平(在外周血检测到或第80天检测到时处死小鼠)。对于所有图形,水平黑线表示平均值,点表示单个动物。**P<0.01,单侧韦尔奇t检验。
图12.AP-1抑制剂 T5224 与VXL协同作用可以一定程度地减轻活体MLLr iB-ALL异种移植的白血病侵袭性。(A)用VXL单独或与T5224抑制剂联合治疗的临床前MLLr iB-ALL PDX模型的体内实验设计。在完成2个基于VXL的化疗周期后1周(第20天),在BM中评估MRD。监测BM、脾脏和外周血的后续复发情况。(B)在治疗前(第0天)确定BM白血病植入,以便根据BM白血病负荷对小鼠进行同样的随机分组,以接受VXL或VXL+T5224治疗。(C)完成2个VXL±T5224周期后1周(第20天),点图监测BM中的MRD。(D)在随访期结束时,白血病小鼠的比例以及BM、脾脏和外周血中的白血病植入水平(在外周血检测到或第80天检测到时处死小鼠)。对于所有图形,水平黑线表示平均值,点表示单个动物。**P<0.01,单侧韦尔奇t检验。
讨论
iB-ALL,尤其是t(4;11)/MLL-AF4+表达的iB-ALL,是儿童时期的一种异常急性白血病,其特征是临床表现早,对当前治疗反应有限,预后不佳。iB-ALL是一种成熟的发育性癌症,在这种癌症中,遗传/分子改变的重新启动是在产前发生的。尽管改进了转基因小鼠和人类异种移植模型,但真实地再现MLLr iB-ALL疾病的潜伏期和表型的历史挑战阻碍了临床/治疗进展。重要的是,最近对iB-ALL进行的全基因组DNA序列研究都揭示了一种基因沉默突变图谱,而不管MLL状态如何。这与该病的极短的潜伏期一起表明,MLLr可能足以导致白血病的发生,或者iB-ALL可能需要极少的DNA协同突变才能导致明确的白血病。MLLr发生的起源细胞和下游转化的造血干细胞和先代细胞(HSPC)能否引发白血病并促进疾病进展,这两个问题仍然是一个争论的焦点。此外,在没有DNA突变的情况下,需要其他非遗传致癌损伤来启动新生儿这种侵袭性白血病的发生。
在这项研究中,研究者对这种突变沉默的iBCP-ALL的DNA甲基组图谱进行全基因组解构可能会为疾病的病理生物学提供独特的见解。计算能力和二代测序方法的最新进展对于揭示其他B细胞恶性肿瘤(包括成人B-ALL、弥漫性大B细胞淋巴瘤、慢性淋巴细胞白血病(CLL)和套细胞淋巴瘤)的基因组、表观遗传组和转录组之间的复杂关系至关重要。在此,本研究整合了69例根据干扰素-99/06方案统一治疗的MLLr和非MLLr iB-ALL白血病患者的全基因组DNA甲基化和转录组的分析数据,以确定全DNA甲基化在iB-ALL发病机制中的作用。
研究报道了iB-ALL样本中的整体DNA低甲基化,类似于在许多人类肿瘤中发现的低甲基化。这一点得到了最近一项全基因组甲基组研究的支持,该研究对具有一致性iB-ALL的白血病双生子进行了研究,该双生子表现出惊人的相似的全基因组甲基组图谱,支持一致性iB-ALL的聚合基因进化。值得注意的是,在iB-ALL中发现的表观遗传中的明显异常的与在自然发生的B细胞成熟的过程中观察到的变化一致。这与以前在CLL和其他B细胞肿瘤中的发现一致,表明在生理过程中发生的受调控的DNA甲基化变化,如B细胞分化,会干扰真正的癌症特异性甲基化变化的发现。事实上,通过分析健康的BCP和原始B细胞,可以将iB-ALL亚型的甲基组区分开来,因此,选择了一种“过滤”策略,在这种策略中,正常B细胞分化过程中自然发生的甲基化变化被去除,从而定义了iB-ALL特异性甲基化特征。
白血病患者的诊断和治疗的一个关键方面在于确定所定义的白血病亚型之间的共同和特异性改变。本研究观察到,虽然MLL-AF4+和非MLLr白血病的异常DNA甲基化图谱存在显著差异,但MLL-AF9+白血病与MLL-AF4+和非MLLR亚型均相关。尽管本研究中包含的MLL-AF9+案例数量有限,但该观察结果表明,MLL-AF9+iB-ALL可能显示出共同的异常特征,并且在这种情况下,MLL重排的贡献可能仅占MLL-AF9+iB-ALL中观察到的异常情况的一小部分。这些结果与Stumpel及其同事发现的,在MLL-AF4+和MLL-AF9+患者之间观察到启动子区域的差异化的DNA超甲基化一致。
尽管人类甲基化EPIC平台存在固有的局限性,但本研究揭示了MLLr iB-ALL中异常DNA甲基化与健康造血细胞增强子和转录控制相关的染色质状态之间的潜在关联,表明iB-ALL中对正常造血起关键作用的转录程序的有序分子内稳态在表观遗传学上被破坏。MLL蛋白是一种H3K4 HMT,发现于COMPASS样复合物中,调节RNA–pol Ⅱ–介导的MLL靶基因的转录起始。因此,观察到的MLL靶模体的甲基组改变可能是iB-ALL细胞中MLL活性改变的结果,这与最近一项报告儿童MLLr B-ALL增强子元件DNA低甲基化的研究结果一致。此外,Prange及其同事最近的一项研究提出,MLL-AF4+和MLL-AF9+融合蛋白可以结合到一个独特的增强子库,从而支持这些白血病亚型中DNA甲基化模式改变的不同分布。
由于已知的DNA甲基转移酶(DNMT)没有一种具有超出CpG位点的明确序列特点,因此必须通过其他机制建立癌症中异常但确定的DNA甲基化模式。其中,由组蛋白修饰或DNA结合TFs的立体效应介导的特定位点上的DNA甲基化的缺失可能会损害这些区域正确的DNMT募集,因此可能在重组iB-ALL的甲基化状态中发挥重要作用。在这种情况下,H3K4修饰和DNA甲基化之间的互斥关系可能解释我们分析中观察到的H3K4me3区域的差异富集。DNMT3L介导的从头甲基化最好是在非甲基化H3K4背景下进行,但在H3K4me存在的情况下,DNMT3L的结合受阻,这表明H3K4甲基化可能阻止特定基因组区域的从头DNA甲基化。由于DNA甲基化变化被认为是与TF结合位点缺失相一致的晚期事件,本研究DNA甲基化结果可能反映了多个TF(包括但不限于MLLr)的重新定位导致的表观遗传的重组。对iB-ALL样本中的多个组蛋白标记进行专门的表观遗传学分析,对于进一步阐明iB-ALL中DNA甲基化、组蛋白修饰和染色质可及性之间的复杂关系具有极其重要的意义。
iB-ALL的全面转录组分析结果显示转录程序明显下调。值得注意的是,差异基因表达分析显示MLL-AF4+iB-ALL表现出更大的表达变异性,表明MLL-AF4+iB-ALL患者中存在不同的分子亚群。在这种情况下,各自融合的AF4-MLL+,仅在t(4;11)+iB-ALL患者被证明,需要预先表达HOXA簇基因,并与MLL-AF4合作促进血内皮前体的出现。重要的是,AF4-MLL/HOXA的表达是一个独立的预后因素,可通过总体生存率和无事件生存率均有改善来识别t(4;11)+iB-ALL患者。
基于网络的共表达分析显示MLLr iB-ALL亚型中存在差异改变的主要分子枢纽。研究者观察到,TFs的AP-1家族成员FOS和JUN在MLLr iB-ALL中,尤其是在MLL-AF4+患者中,在建立特定转录特征方面发挥了关键作用。事实上,AP-1复合体的成分在多种人类肿瘤中有着广泛的作用,最近的一项研究表明FOS/JUN相关蛋白在急性髓系白血病(AML)的发病机制中起着至关重要的作用。进一步整合DNA甲基化和基因表达数据,并通过多种正交方法进行广泛验证,证实FOS/JUN蛋白不仅在iB-ALL中异常表达,而且在重塑iB-ALL中的DNA甲基化图谱中发挥关键作用,表明转录因子表达和确定的表观遗传程序的建立之间存在复杂的关系,在这种疾病的背景下,它们尤其不平衡。此外,综合策略能够通过将转录因子的甲基化状态/表达与其下游靶基因之间的功能结果联系起来,从而明确特定的调控网络,发现iB-ALL中癌细胞脆弱性。
多种表观遗传和转录组事件扰乱了正常B细胞分化所需的调节网络。对MLLr iB-ALL中下调分子枢纽(包括AP-1轴)的特征和功能进行体外和体内验证,为更好地理解MLLr iB-ALL的启动和发病进程提供了新的见解,通过靶向方法的组合,为MLLr iB-ALL开辟了潜在的治疗途径。未来的研究应该解决通过全基因组整合甲基组转录组分析描述的癌细胞脆弱性,以及针对MLL iB-ALL中AP-1成员的潜在治疗机会是否可以扩展到成人MLLr B-ALL和MLLr AML。
原始出处:
Tejedor JR, Bueno C, Vinyoles M, Petazzi P, Agraz-Doblas A, Cobo I, Torres-Ruiz R, Bayón GF, Pérez RF, López-Tamargo S, Gutierrez-Agüera F, Santamarina-Ojeda P, Ramírez-Orellana M, Bardini M, Cazzaniga G, Ballerini P, Schneider P, Stam RW, Varela I, Fraga MF, Fernández AF, Menéndez P.Integrative methylome-transcriptome analysis unravels cancer cell vulnerabilities in infant MLL-rearranged B cell acute lymphoblastic leukemia.J Clin Invest. 2021 Jul 1;131(13):e138833. doi: 10.1172/JCI138833

