病例报告|伴白质病变的强直性肌营养不良的家系1例
时间:2025-01-22 12:10:39 热度:37.1℃ 作者:网络
摘 要 强直性肌营养不良1型(myotonic dystrophy,DM1)是以肌强直和肌无力为突出症状的罕见遗传病,可累及全身多个系统。本文报告1例中枢神经系统症状轻微,影像学检查已出现明显脑白质病变的DM1病例,以提高对DM1累及神经系统的认识。患者为48岁男性,以肌无力、肌萎缩为主要表现,伴轻微记忆力下降,肌酸肌酶升高,肌电图呈肌源性改变、肌强直电位,头颅磁共振检查示多发脑白质病变,基因检测确诊DM1,予营养神经、改善认知等治疗,随访1年病情稳定。
关键词
肌营养不良;肌强直;肌无力;脑白质病变;CADASIL;基因检测;MRI
强直性肌营养不良1型(myotonic dystrophy type 1, DM1)是一种常染色体遗传病,以肌强直、进行性肌无力为特征。该病常累及全身多个系统,临床表现复杂多样。其中,中枢神经系统(central nervous system, CNS)症状可表现为疲劳、白天嗜睡、抑郁、癫痫、认知功能障碍等[1-3],早期容易被忽视,或因其与其他疾病重叠而导致误诊。本文报告1例临床未伴有明显认知功能障碍、癫痫等CNS症状,影像学检查已出现明显脑白质病变的DM1家系,分析其临床表现、诊疗情况,以提高对DM1累及神经系统的认识。
1 临床资料
1.1 先证者 男,48岁,打杂工,因“四肢肌肉萎缩10余年,加重伴发音含糊3年”于2022年4月15日入院。患者于10多年前无明显诱因下出现四肢远端肌肉萎缩伴乏力,以双下肢为著,伴握拳后放松困难,反复握拳症状可减轻,3年前症状逐渐明显,行走欠稳健,出现发音含糊、进食呛咳,伴轻微记忆力下降,无抽搐、头晕头痛、肉跳感、胸闷心悸、呼吸困难、腹胀、呕吐、反酸等,二便正常。既往19年前有肺结核、脊柱结核,行手术治疗,术后服用抗结核药物,已停药,有白内障病史。否认烟酒嗜好及毒物接触史。内科查体无明显异常。神经科查体:神清,秃顶,斧状脸,对答切题,双瞳孔正圆等大,对光反射灵敏,双眼外斜位,发音含糊,双侧大小鱼际肌稍萎缩,双小腿肌肉明显萎缩(图1),叩击虎口肌肉出现肌强直,双上肢近端肌力5级,握力4-级,双下肢近端肌力5级,远端肌力4级,肌张力稍低,四肢腱反射减退,病理征未引出。查肌酸激酶513.00 U/L(参考值0~174 U/L);催乳素368.78 μIU/mL(参考值55.97~278.36 μIU/mL);甲胎蛋白7.56 IU/mL(参考值0~5.8 IU/mL),细胞蛋白19片段3.51 ng/mL(参考值0~3.3 ng/mL),神经元特异性烯醇化酶19.51 ng/mL(参考值0~16.3 ng/mL);血常规、二便常规、血沉、C反应蛋白、乳酸、肝肾功能、代谢八项、甲状腺功能、止凝血、传染病八项、风湿三项、免疫五项、抗核抗体谱均正常。蒙特利尔认知评估量表(Montreal cognitive assessment, MoCA)28分。心电图、心脏彩超未见明显异常。头颅MRI(图2):双侧半卵圆中心、侧脑室旁白质及放射冠散在多发异常信号,左侧颞极皮质及皮质下白质、右侧颞极白质异常信号灶,性质待定,脑小血管病?遗传性脑血管病?脱髓鞘病变?肌电图:四肢多发慢性肌源性损害(广泛类似轰炸机俯冲声音的肌强直电位,远端肌肉为著)。强直性肌营养不良蛋白激酶(dystrophia myotonic protein kinase, DMPK)基因3'UTR区域CTG重复数目一个正常(拷贝数4),一个发生异常扩增(拷贝数>100),符合DM1的基因突变特征。
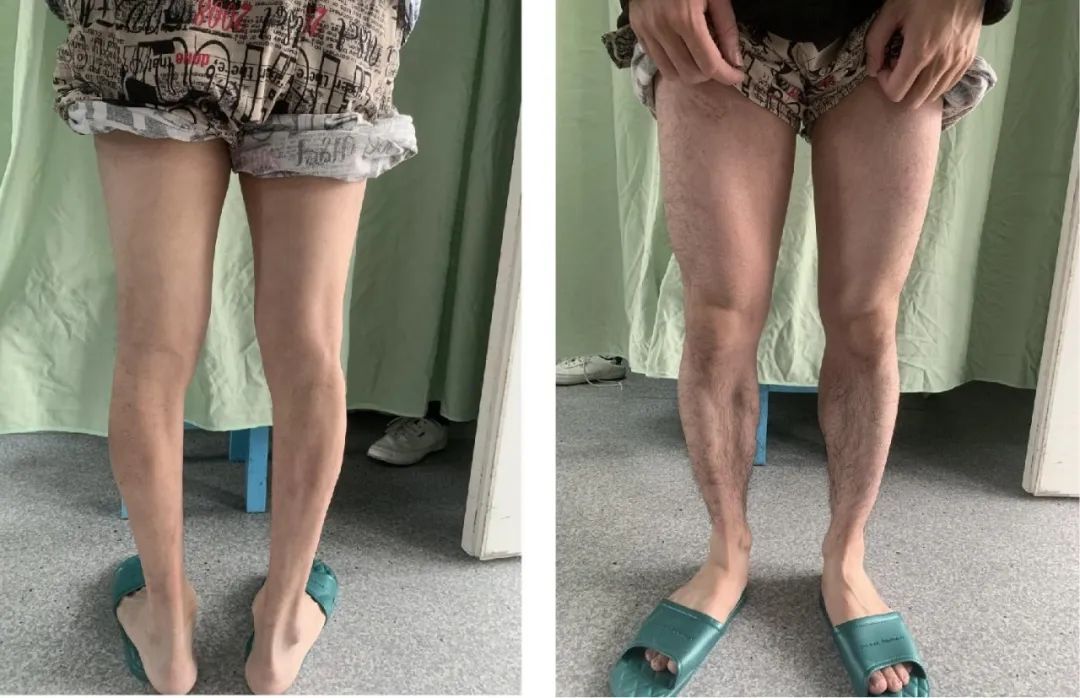
图1 先证者下肢肌肉萎缩Fig.1 Atrophy of the lower limb muscles in the proband
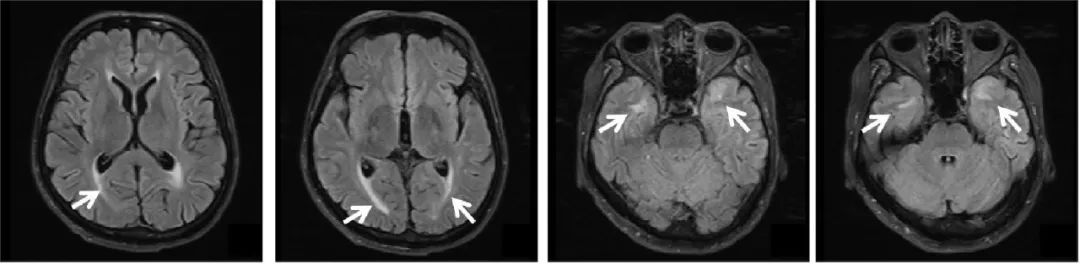
图2 先证者头颅MRI 双侧侧脑室旁白质及放射冠散在多发T2-Flair高信号,左侧颞极皮质及皮质下白质、右侧颞极白质T2-Flair高信号,如箭头所示。Fig.2 Magnetic resonance imaging of the proband’s cranium
1.2 家系调查 见家谱图(图3)。Ⅲ 1为先证者大儿子,23岁,约10年前开始出现四肢乏力,伴发音含糊,伴左上肢肌肉萎缩,无进食呛咳、胸闷、呼吸困难、腹胀、恶心、反酸、记忆力下降,二便正常。否认烟酒嗜好及毒物接触史。查体:内科查体无明显异常,神清,发音稍含糊,左侧大小鱼际肌萎缩,四肢近端肌力5级,远端肌力4级,病理征未引出。2022年4月18日我院查肌酸激酶424 U/L;血常规、肝肾功能、血糖、甲状腺功能均正常。心电图、头颅MRI未见异常。肌电图:四肢多发肌源性损害,呈广泛肌强直放电。基因检测示DMPK基因3'UTR区域CTG重复数目发生异常扩增(拷贝数>50)。
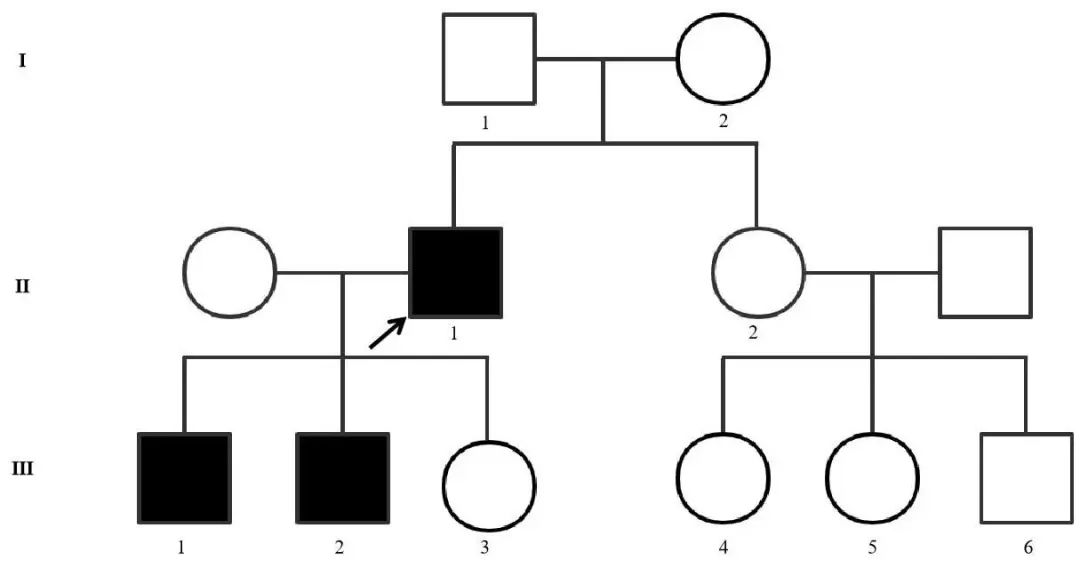
图3 强直性肌营养不良的家系图谱 □为未患病男性,○为未患病女性,■为患病男性,↗为先证者。Fig.3 Pedigree chart of myotonic dystrophy in this case
Ⅲ 2为先证者小儿子,22岁,约10年前开始出现四肢乏力,伴发音含糊,逐渐加重,讲话难以听清,无肌肉萎缩、进食呛咳、胸闷、呼吸困难、腹胀、恶心、反酸、记忆力下降,二便正常。否认烟酒嗜好及毒物接触史。查体:内科查体无明显异常,神清,发音含糊,无明显肌肉萎缩,四肢近端肌力5级,远端肌力4级,病理征未引出。2022年4月18日我院查肌酸激酶740 U/L,超敏促甲状腺素7.305 μIU/mL(参考值0.38~5.33 μIU/mL);血常规、血糖、肝肾功能均正常。心电图、头颅MRI未见异常。肌电图:四肢多发肌源性损害,呈广泛肌强直放电。基因检测:DMPK基因3'UTR区域CTG重复数目发生异常扩增(拷贝数>50)。
家系中患病者临床确诊DM1,给予营养神经、指导康复锻炼,症状无明显改善。随访1年,患病者病情无明显变化。
2 讨论
DM1是染色体19q13.3上DMPK基因3'端非翻译区CTG重复异常扩增导致[4]。DM1常因累及肌肉系统出现肌萎缩及肌强直。本家系中3例患者均以肌无力起病,四肢远端受累明显,肌酶升高及肌电图肌源性损害需与其他肌病鉴别。既往文献指出,出现肌强直表现及肌电图产生类似轰炸机俯冲声音的肌强直电位为重要的鉴别点[5-7]。本例家系符合上述特点,并通过基因检测确诊。先证者还出现双侧面部无力和轻度眼睑下垂,呈现“斧状脸”的特征性面容。DM1还可导致早发性白内障、甲状腺功能异常、糖尿病、心脏疾病等,其中心律失常和呼吸衰竭是常见的死因[5,8]。此外,本例先证者数个肿瘤指标增高。有文献指出DM1患恶性肿瘤风险增高[9]。因此,建议DM1患者针对各个系统定期体检,以及时治疗,减少并发症的发生,提高患者生活质量。
以往报道的DM1病例多关注患者典型的肌肉系统症状,CNS受累因早期症状不明显容易被忽视。DM1可出现疲劳、白天过度嗜睡、抑郁、癫痫、认知功能障碍等[1-3],影响了患者的生活质量,其中有一些症状是可干预的。影像学上患者CNS可呈现脑白质病变、脑萎缩、脑室扩张、血管间隙增宽,其中脑白质病变以侧脑室周围白质、额颞叶受累多见[2-3]。累及岛叶者需与自身免疫性脑炎鉴别,脑脊液检查及抗体检测可协助明确[2]。研究还发现,DM1脑白质高信号并不累及丘脑、基底节区[2]。本例先证者出现轻微记忆力下降,头颅MRI出现双侧颞极病变,影像学上与伴有皮质下梗死和白质病变的常染色体显性遗传性脑动脉病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL)难以鉴别,容易误诊[10]。脑卒中病史、磁敏感多发微出血的影像改变可能提示CADASIL[11],“斧状脸”的特殊面容及肌强直电位则有助于诊断DM1[5],基因检测可帮助确诊。既往报告的伴脑白质病变的DM1病例多为已经合并严重认知功能障碍[1]或癫痫[2]等CNS症状的患者。本例先证者主诉轻微的记忆力下降,客观的MoCA评分未发现明显认知功能损害,而影像学检查已出现明显脑白质病变,提示DM1的CNS受累可能在疾病更早期已出现,临床工作中应予以关注。Ⅲ1、Ⅲ2目前无CNS症状及影像学改变,仍需随访。
综上,DM1临床表现的复杂性、多样性及与其他疾病的重叠性,给临床确诊带来挑战,充分认识DM1在不同系统的临床表现,对早期识别疾病十分重要。其中,CNS受累在早期难以发现且更容易误诊,建议MRI检查可作为DM1的常规检查项目,有利于帮助早期识别、积极干预,以提高患者的生存率及生活质量。
参考文献:
1. 李楠, 张续, 苏春贺, 等. 伴脑白质病变的强直性肌营养不良1例报告[J]. 中国实用神经疾病杂志, 2020, 23(21): 1923-1927.
2. 梁欣, 刘兰祥. 伴脑白质病变的强直性肌营养不良症两例[J]. 磁共振成像, 2022, 13(2): 101-102.
3. VAN DORST M, OKKERSEN K, KESSELS R P C, et al. Structural white matter networks in myotonic dystrophy type 1[J]. Neuroimage Clin, 2019, 21: 101615.
4. DE PONTUAL L, TOME S. Overview of the complex relationship between epigenetics markers, CTG repeat instability and symptoms in myotonic dystrophy type 1[J]. Int J Mol Sci, 2022, 23(7): 3477.
5. JOHNSON N E. Myotonic muscular dystrophies[J]. Continuum (Minneap Minn), 2019, 25(6): 1682-1695.
6. 向栩莹, 孙洲, 陈晓露, 等. 强直性肌营养不良1型肌肉症状与特征性血清学指标分析[J]. 中国神经精神疾病杂志, 2020, 46(8): 449-454.
7. 贾妮, 赵丽云. 强直性肌营养不良的分子基因学研究进展[J]. 神经疾病与精神卫生, 2004, 4(6): 475-476.
8. 徐玲, 王方芳, 冯新恒, 等. 强直性肌营养不良6例心脏表现及文献复习[J]. 中国循环杂志, 2022, 37(7): 726-730.
9. EMPARANZA J I, LÓPEZ DE MUNAIN A, GREENE M H, et al. Cancer phenotype in myotonic dystrophy patients: Results from a meta-analysis[J]. Muscle Nerve, 2018, 58(4): 517-522.
10. 吴昊天, 张炳俊, 杨渝, 等. 类似CADASIL的1型强直性肌营养不良一例[J]. 中国神经免疫学和神经病学杂志, 2020, 27(4): 334-335.
11. ZHU S, NAHAS S J. CADASIL: Imaging characteristics and clinical correlation[J]. Curr Pain Headache Rep, 2016, 20(10): 57.
【引用格式】陈俊玲,黄林欢,吴楚钟,等. 伴白质病变的强直性肌营养不良的家系1例[J]. 中国神经精神疾病杂志,2024,50(11):678-680.
【Cite this article】CHEN J L,HUANG L H,WU C Z,et al.A case of myotonic dystrophy with white matter lesions[J]. Chin J Nervous Mental Dis,2024,50(11):678-680.
DOI:10.3969/j.issn.1002-0152.2024.011.007

