T细胞活化和检查点抑制中的泛素化
时间:2022-10-13 16:03:21 热度:37.1℃ 作者:网络
前言
基于T细胞的免疫疗法的出现显著改变了癌症患者的治疗模式。尽管取得了成功,但目前批准的免疫治疗方案仍然存在局限性。因此,需要对T细胞活化和抑制的分子机制有更深入的了解,以合理地扩大改善免疫治疗的靶点和可能性。
免疫信号通路下游的蛋白质泛素化对于几乎所有免疫反应,特别是T细胞激活的正调节和负调节至关重要。大量研究表明,泛素化通过控制蛋白质的功能、定位和稳定性发挥多种细胞和分子作用,对泛素依赖性通路的调节可以显著改变T细胞的活化并增强抗肿瘤反应。因此,人们正在积极开发有选择性地利用泛素相关酶用于癌症治疗的技术,靶向泛素化为推进T细胞免疫治疗提供了巨大的可能性。
蛋白质泛素化
泛素化是一种翻译后修饰,泛素是一种由76个氨基酸组成的小蛋白质,通常由一条泛素链与底物蛋白质内的赖氨酸残基共价连接。泛素化通过各种方式影响蛋白质功能,例如通过影响蛋白质稳定性、周转率、细胞定位,以及诱导影响与其他蛋白质相互作用的构象变化。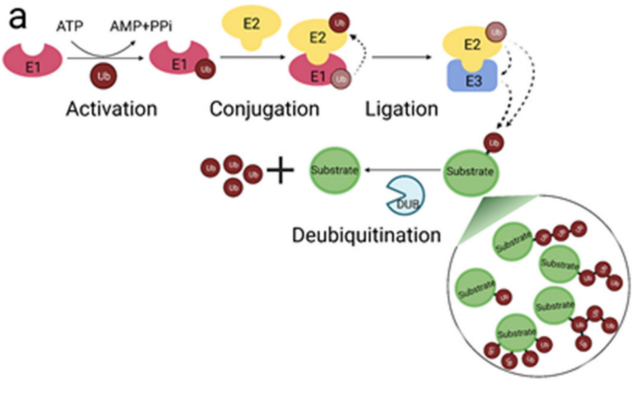
该过程由E1泛素激活酶启动,该酶使用ATP激活,然后将泛素单体转移到E2泛素结合酶。泛素最终在E3泛素连接酶的作用下从E2转移到底物。
泛素化是多方面的,底物蛋白可以仅用一个泛素(单泛素化)、多个单泛素(多个单泛素化)或多泛素链(多泛素化)标记。由于泛素本身有七个赖氨酸(Lys6、Lys11、Lys27、Lys29、Lys33、Lys48和Lys63)和一个N端甲硫氨酸残基(M1),可以进行泛素化,因此可以形成几种结构不同的同质/同型、混合/异型和分支多泛素链。

泛素化除了是通过蛋白质体降解蛋白质的主要调节器外,实际上是几乎所有细胞信号级联的多功能调节器。泛素化蛋白质的命运取决于添加的泛素数量和形成泛素连接的泛素氨基酸。通过这种方式,并且由于拓扑结构不同链的多种可能性,泛素构成了一种高度通用的信号。
单泛素化和多泛素化参与膜受体的内吞作用,以及蛋白质降解、定位和蛋白质-蛋白质相互作用,这些类型的链也与TGF-β信号传导有关。在同型多泛素链中, Lys48和Lys63连接的链是最丰富,它们分别驱动蛋白酶体降解和调节多种细胞信号事件。Lys11连接链调节细胞周期和线粒体自噬,M1连接链对炎症固有反应至关重要,特别是调节NF-κB信号。
由Lys6-、Lys27-、Lys29-、Lys33泛素连接链调节的细胞过程仍然不太清楚。Lys27连接链似乎对调节先天免疫、细胞周期和线粒体功能非常重要。Lys6链似乎参与DNA修复和线粒体自噬,而Lys29链与Wnt信号以及蛋白毒性应激和细胞周期相关。Lys33链在高尔基体后转运和T细胞受体(TCR)调节中起着关键作用。
此外,非常重要的是,泛素在底物上的组装可以通过去泛素化酶(DUB)的作用逆转,去泛素化酶水解并从底物上去除泛素,使泛素化成为一种瞬时修饰,DUB对于维持生理性泛素平衡是必不可少的。这种平衡的失调与人类疾病的发病机制有关,特别是癌症、感染、神经退行性变和免疫紊乱,因此,DUB已开始作为基于抑制剂的治疗成为有吸引力的药物靶点。
TCR/CD3激活中的泛素途径
E3连接酶
E3泛素连接酶Casitas B系淋巴瘤(Cbl-B)是最具特征的E3连接酶,是T细胞活化的主要参与者。在T细胞激活时,它在免疫突触处被招募到TCR中,在TCR下游发挥多种抑制机制。
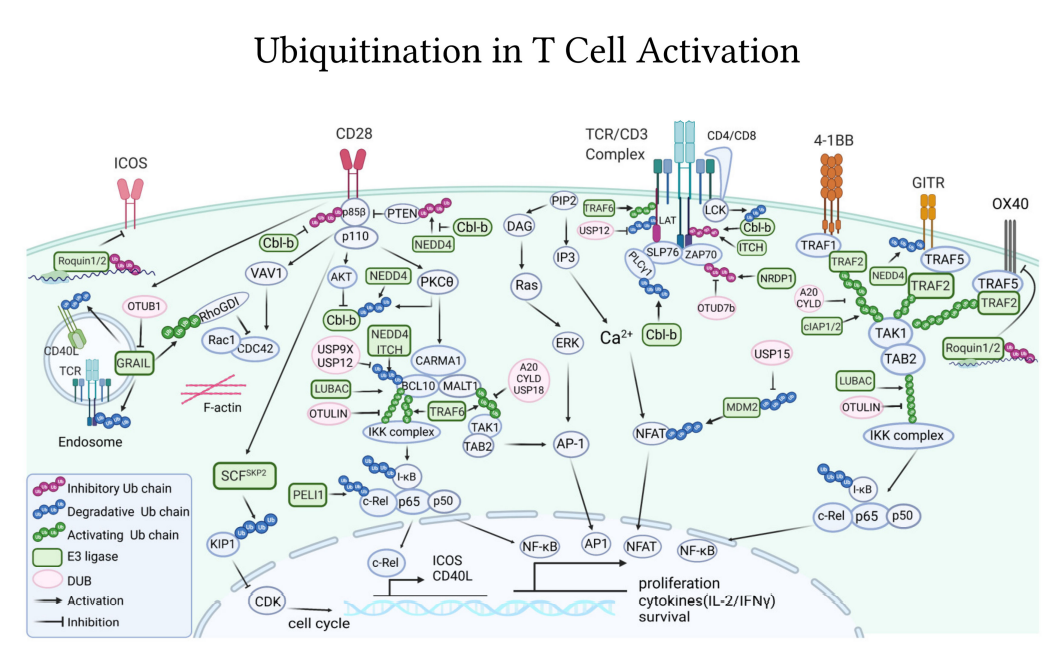
Cbl-b通过其多个蛋白质相互作用结构域与关键TCR信号体分子(如LCK、SLP76、ZAP70、Vav1、PKCθ和PI3K)相互作用。Cbl-b与E3连接酶Itch协同作用,能够介导TCR-ζ的Lys33连接的多泛素化,其不针对TCR受体进行降解或内吞,而是阻止其磷酸化并进一步与下游ZAP70激酶结合。通过这些相互作用,以及一些信号体成分的泛素化,Cbl-b强烈抑制T细胞活化。Cbl-b敲除小鼠具有过度活跃的T细胞,其激活不需要CD28,此外,Cbl-b基因敲除小鼠中从未描述过自身免疫损伤。因此,Cbl-b具有多个检查点抑制作用和极小的自身免疫毒性,是未来靶向癌症免疫治疗的有力候选。
与淋巴细胞无能相关的基因(GRAIL)是一种跨膜E3连接酶,定位于内体,也对T细胞活化起负调节作用。TCR介导的T细胞活化促进GRAIL的表达,而GRAIL反过来又阻碍T细胞活化。机制上,GRAIL通过非降解泛素链稳定和激活Rho-鸟嘌呤解离抑制剂(RhoGDI),导致细胞骨架重排和IL-2分泌缺陷。
NRDP1是另一种损害TCR信号的E3连接酶。T细胞激活后,NRDP1将Lys33多聚泛素非降解链添加到ZAP70激酶。Lys33连接的泛素链有助于招募Sts1和Sts2磷酸酶,最终使ZAP70去磷酸化和失活。
除了作用于近端信号体外,E3连接酶还通过进一步作用于下游抑制T细胞活化,控制导致T细胞活化转录因子核易位的关键转导事件。例如,在TCR/CD28连接后,E3连接酶Pellino1(PELI1)通过用Lys48多泛素链标记c-Rel来阻止NF-κB通路,导致其蛋白酶体降解。
DUBs
去泛素化酶A20和CYLD,在调节天然免疫细胞中的NF-κB信号中具有重要作用,也能显著改变T细胞功能。外周T细胞中A20的条件性缺失导致CD8+T细胞具有增强的NF-κB信号,即使在低抗原剂量下也能更有效地被激活,以产生更高水平的IL-2和IFN-γ。
CYLD可以通过NF-κB损害T细胞活化,它是通过将CBM复合物下游TAK1激酶的Lys63泛素链进行去泛素化。CYLD缺陷小鼠出现结肠炎,T细胞频率和活化增加。
使用类似的作用机制,去泛素酶USP18对TAK1的去泛素化抑制TCR。USP18的基因缺失导致T细胞中NF-κB和NFAT的过度激活和IL-2的过度产生。相反,去泛素酶USP9X和USP12通过从BCL10中去除抑制性泛素链来积极调节NF-κB通路,从而阻断CBM信号复合物的组装。因此,缺乏USP9X或USP12的T细胞在TCR激活时表现出较低水平的NF-κB激活,以及增殖和细胞因子生成减少。
TCR内化
TCR信号幅度和持续时间由细胞膜受体的内化、再循环和降解的平衡决定。在抗原刺激下,TCR/CD3复合物是泛素化的,而泛素化对于表面受体的内化至关重要。据报道,Cbl-b可单独或与E3连接酶c-Cbl联合促进TCR的内化,Cbl-b还干扰激活和聚集,从而破坏免疫突触的稳定性,进一步减弱TCR信号。
类似地,E3连接酶GRAIL可以通过蛋白酶体多泛素化和降解CD3ζ分子来下调表面TCR/CD3复合物的表达。最近的研究表明,用于CAR-T治疗的CAR受体的表面表达也受到泛素介导的内吞和降解的影响。注入患者体内的CAR-T细胞持续性差是这种肿瘤免疫疗法的主要限制。最近的一项研究能够通过对细胞质结构域的所有赖氨酸残基进行突变来提高CAR的稳定性,这有效地绕过了CARs的泛素化依赖性溶酶体降解,因此,这些CAR-T细胞改善了CAR的持久性和来自内体的信号传导,同时增强了效应器功能,从而在小鼠模型中产生持久的抗肿瘤免疫反应。
共刺激受体下游的泛素途径
CD28
E3连接酶Cbl-b是CD28介导的信号传导的主要抑制剂,从机制上讲,E3连接酶Cbl-b通过结合和泛素化PI3K的p85β亚单位来抑制CD28依赖的PI3K途径。
E3连接酶不仅能阻碍CD28信号传导,而且能促进CD28信号传导。例如,TCR/CD28激活NFAT涉及E3连接酶TRAF6。TRAF6被招募到免疫突触与支架蛋白LAT相互作用,并通过Lys63泛素化增强LAT磷酸化和TCR信号。
为了充分激活T细胞,CD28共受体必须克服Cbl-b抑制。CD28信号通过触发Cbl-b的翻译后修饰来阻断Cbl-b抑制途径,最终导致其蛋白酶体降解。其中E3连接酶NEDD4发挥着重要作用,据报道NEDD4在CD28共刺激下结合并泛素化Cbl-b进行蛋白酶体降解。
其他共刺激受体
与CD28类似,肿瘤坏死因子受体(TNFR)家族成员的共刺激受体4-1BB、CD40L、OX40和GITR与TCR协同促进T细胞活化,尤其是增殖和细胞因子的产生。作为TNFRs,它们缺乏内在的酶功能,依赖于几种E3连接酶,包括TRAF(TRAF1/2/3)、cIAP1/2和LUBAC,在导致NF-κB活化的信号转导级联中引导大量Lys63、Lys48和线性泛素化事件。去泛素酶A20、CYLD和OTULIN可以清除这些泛素化事件以阻止NF-κB的活化。
抑制性受体下游泛素途径
CTLA-4
到目前为止,没有任何E3直接泛素化CTLA-4的报道,也没有CTLA-4中泛素化位点的记录。然而,CTLA-4缺陷型T细胞的泛素修饰总体上显著减少,这有力地表明了泛素化事件在CTLA-4途径中的重要性。此外,实验证据表明,主要CTLA-4抑制功能由关键的T细胞抑制性E3连接酶介导,该连接酶也调节T细胞活化,即Cbl-b、ITCH和GRAIL。

此外,Cbl-b、ITCH和GRAIL是T细胞无能的基本驱动因素,而且这三种E3连接酶还通过调节Treg的发育和功能发挥重要的免疫抑制作用。Cbl-b和ITCH参与TGF-β介导的Foxp3调节,缺乏这两种E3连接酶都会损害TGF-β诱导的Foxp3+Tregs(iTreg)的发育,导致iTreg低表达,功能缺陷。
PD-1
与CTLA-4不同,一些报告强调了泛素化在PD-1/PD-L1系统中的关键作用。对于T细胞上的PD-1受体,Cbl-b也作为PD-1抑制途径的关键介质显著参与,缺乏Cbl-b的T细胞对PD-1抑制具有抵抗力。没有Cbl-b,PD-1在抑制IFN-γ产生或诱导T细胞活化后的细胞死亡方面效率低下。
另一种Cbl E3连接酶c-Cbl也影响PD-1,但与Cbl-b相反,它是一种负性调节因子。c-Cbl与PD-1的胞质尾部结合,并作为E3连接酶,泛素化PD-1进行蛋白酶体降解。因此,c-Cbl减少提高了CD8+T细胞和巨噬细胞中PD-1的表达。
同样,E3连接酶FBXO38通过介导PD-1降解来控制T细胞抗肿瘤反应。从机理上讲,FBXO38通过在Lys233处添加Lys48多聚泛素链来标记PD-1进行降解。缺乏FBXO38的黑色素瘤和结直肠癌小鼠模型具有更高的肿瘤负荷,这与肿瘤浸润性T细胞中PD-1的高表达有关。
PD-L1
PD-L1的肿瘤水平是肿瘤免疫的重要决定因素。几项研究已确定癌细胞利用EGFR信号稳定PD-L1表达以逃避T细胞免疫。E3连接酶识别并泛素标记PD-L1进行蛋白水解,主要依赖于糖原合成酶激酶3(GSK3α/β)。GSK3β磷酸化PD-L1的C端结构域,以招募E3连接酶β-TrCP用于PD-L1泛素依赖性降解。而EGFR信号通过诱导GSK3β磷酸化位点附近的PD-L1大量糖基化来对抗GSK3β/β-TrCP/PD-L1降解途径。
另一项研究进一步表明GSK3α/ARIH1是另一种诱导PD-L1蛋白水解的激酶/E3连接酶对。E3连接酶ARIH1识别S279/S283处PD-L1的GSK3α依赖性磷酸化,随后PD-L1 Lys48泛素化和降解。最近,还发现EGFR通过额外干扰膜结合MARCH8 E3连接酶增加PD-L1水平,该连接酶也以PD-L1为降解靶点。最后,c-Cbl过表达,单独或与Cbl-b联合,也可以降低STAT3/AKT/ERK磷酸化,从而下调PD-L1表达并增加抗肿瘤反应。
Cullin3 SPOP和TRIM21E3连接酶通过泛素化依赖性降解进一步破坏PD-L1的稳定性,它们的作用再次依赖于激酶,特别是细胞周期蛋白依赖性激酶CDK4和CDK5。然而,CDK4通过稳定Cullin3 SPOP协助PD-L1降解,而CDK5则消极干扰TRIM21对PD-L1的作用。
此外,PD-L1也因其他几种去泛素化酶而稳定,即OTUB1、USP9X、USP7。这些研究具有潜在的治疗意义,因为它们证明,抑制或清除这些DUB中的任何一种都可以重新激发小鼠的抗肿瘤反应,并使癌细胞对T细胞杀伤敏感。
泛素依赖途径肿瘤药物的开发
到目前为止,针对泛素依赖的途径主要是基于小分子抑制剂,这些小分子可以通过直接结合或变构作用阻断泛素相关酶的催化结构域,或阻止其与底物或其他调节蛋白结合。目前,已经开发了200多种化合物,其中许多化合物正在进行癌症治疗的临床前和临床试验。
大多数基于泛素的小分子化合物被设计成靶向癌细胞中必需的泛素途径。随着以T细胞为基础的肿瘤免疫治疗的出现,针对选择性E3和DUB的靶向抑制已有研究,其中关键的细胞内检查点T细胞抑制剂Cbl-b具有巨大的潜力。已有研究利用RNA干扰成功地下调了T细胞中的Cbl-b水平,这在不同的过继性T细胞转移肿瘤模型中产生了出色的体外和临床前结果,并且癌症患者对其耐受性良好,目前已进入临床试验(NCT02166255和NCT03087591)。此外,针对IAPs、MDM2和USP7的小分子已经进行了临床前试验,并证明单独使用或联合使用时,至少部分增强体内抗肿瘤免疫。
调节癌细胞中PD-L1降解的泛素化依赖途径也正在进行测试。例如,一种竞争性棕榈酰化抑制剂(CPP-S1),以防止PD-L1棕榈酰化,进而促进PD-L1泛素介导的降解。控制PD-L1泛素化和降解的EGFR信号通路也通过不同的方式在体内被有效阻断,包括EGFR小分子抑制剂、铜螯合剂和靶向CSN5的小分子,都显著增强T细胞抗肿瘤反应。
基于邻近性的治疗方法,包括PROTAC或分子胶技术,也正在迅速发展,以针对几种特定肿瘤蛋白质的蛋白酶体降解,目前有十几项临床试验正在验证这些基于泛素化技术的治疗潜力。
小结
数据表明,调节几种泛素依赖性途径,特别是所涉及的泛素依赖性酶,可以释放强烈、持久和靶向的抗肿瘤反应。目前,泛素领域也取得了巨大进展,开发出了多种新技术和新方法,可以专门针对几乎所有泛素依赖的酶,或者利用它们针对感兴趣的底物蛋白,包括肿瘤蛋白。展望未来,相信这些重要的泛素依赖途径的调节很快将成为改善靶向肿瘤免疫治疗的可行替代方案。
: , 。 视频 小程序 赞 ,轻点两下取消赞 在看 ,轻点两下取消在看

